ICML 2025 Preview


ICML 2025
Every summer, the season for ICML, one of the top three AI conferences, approaches. This year, The 42nd International Conference on Machine Learning (ICML 2025) is scheduled from July 13 to July 19. As I write this, we are awaiting the submission of camera-ready versions (fully revised and polished manuscripts for publication) of accepted papers. However, the list of accepted papers has already been released, so I’ve decided to look at the papers relevant to our field. Our AI research team takes turns previewing AI conferences, and coincidentally, I’m covering ICML again this year, just like last year 😄.
Let’s start with some statistics about ICML papers. It’s widely agreed that AI is not a fleeting trend but a lasting force, continuing to dominate various industries and even government policies. This is reflected in the number of submissions and accepted papers at AI conferences like ICML. The number of submissions to ICML has surged over the past two years. Last year, there were 9,653 submissions, and this year, the number jumped to 12,107—a remarkable 25% increase. [Paper Copilot].
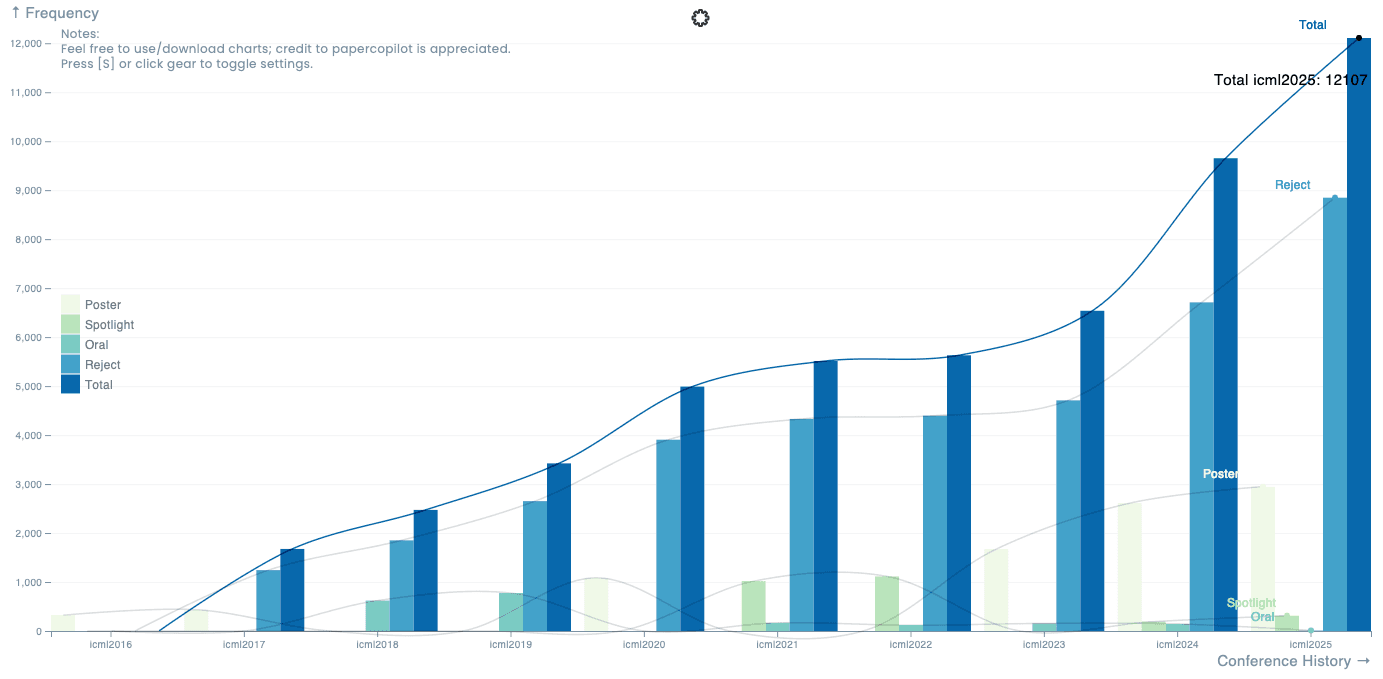
Meanwhile, the number of papers accepted for presentation—including posters, spotlights, and oral sessions—was 3,260 (or 3,340 according to the ICML website), resulting in an acceptance rate of 27%. This is slightly lower than last year’s rate of 31%. While more research is being conducted overall, the proportion of papers accepted for conference presentations appears to be consistently regulated. Acceptance rates at other major AI conferences vary slightly but remain in a similar range: 23% at AAAI 2025, 32% at ICLR 2025, and 26% at NeurIPS 2024.

ICML 2025: Papers Related to Drug Development
For this preview, I’ve compiled a list of papers related to drug development from those accepted for presentation at the conference. These papers were gathered by searching for keywords such as chem, drug, and mol on the official paper list page.
This year, the number has surpassed 100. This increase, beyond the overall rise in total submissions, likely reflects the growing interest within the AI community in applying AI to (natural) science problems, often referred to as AI4Science.
You can find the curated list of papers below. Please note that some papers loosely related to drug development, such as those in computational chemistry or materials science, are also included, so keep that in mind 😉
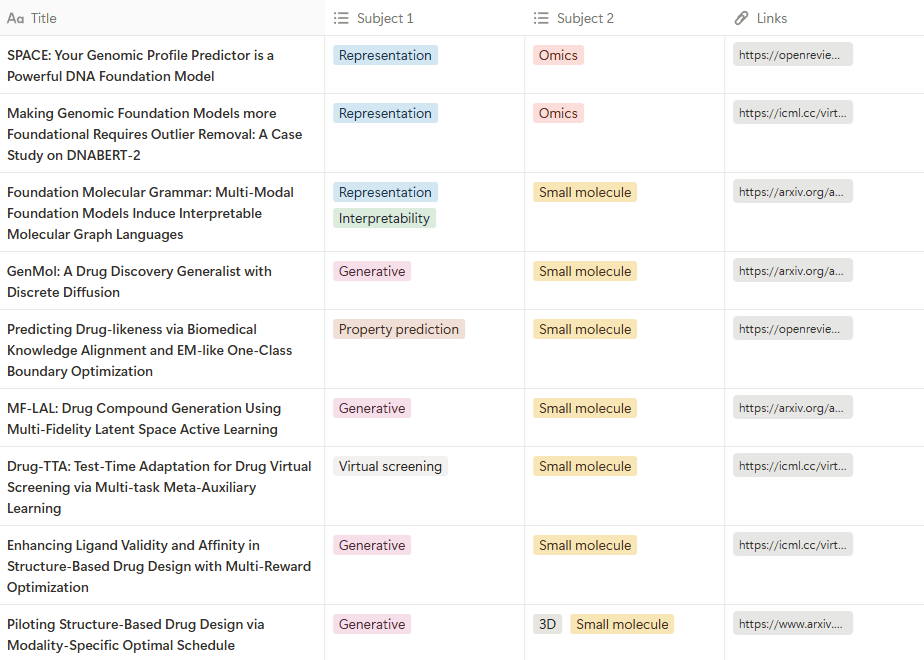
👉 Click the image to visit the page.
As the number of relevant papers has increased overall, the range of topics covered has also become more diverse. While generative model studies for designing small ligands or proteins remain prominent, bioinformatics topics dealing with various omics data have noticeably grown. Additionally, unlike molecules such as proteins, generative model research for crystalline structures, which require unique characteristics like symmetry and periodicity, has seen a significant rise.
Next, I’ll take a closer look at some of the research being presented at ICML 2025, using the papers listed below as representative examples.
- Shen, Seo, et al. Compositional flows for 3D molecule and synthesis pathway co-design [ICML 2025].
- Wu, Padia et al. Identifying biological perturbation targets through causal differential networks [ICML 2025].
- Bendidi, El Mesbahi, et al. A cross-modal knowledge distillation & data augmentation recipe for improving transcriptomics representations through morphological features [ICML 2025].
- Joshi, Fu et al. All-atom diffusion transformers [ICML 2025].
ICML 2025 Preview 1 | 3DSynthFlow: Co-design of 3D Molecular Structures and Synthesis Pathways
The first paper we’ll explore is “Compositional flows for 3D molecule and synthesis pathway co-design” by Tony Shen, Seonghwan Seo, and colleagues. In molecular design using generative AI, most approaches focus either on the 3D structure of a molecule (e.g., its binding conformation to a target protein) or its synthetic feasibility, often resulting in suboptimal performance in the other aspect.
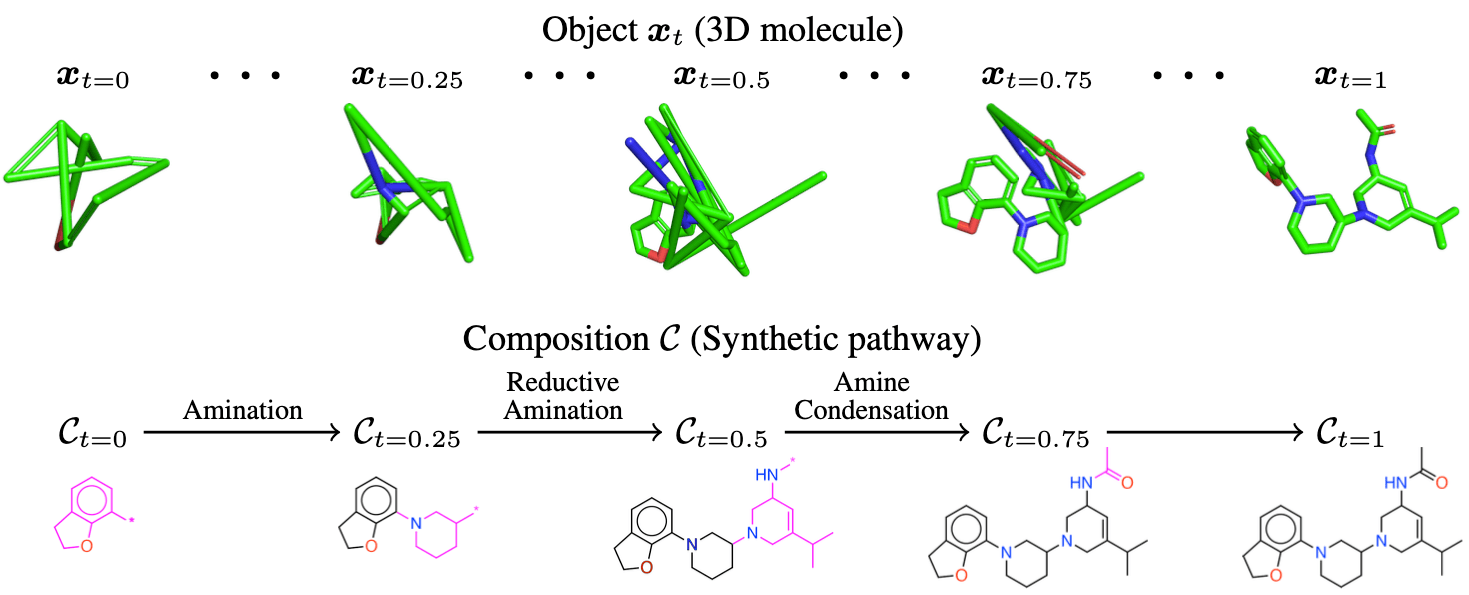
This paper addresses both challenges simultaneously by representing a molecule in terms of its composition and state of matter. Here, the composition refers to how the molecular structure is formed by connecting specific fragments, reflecting the process where substructures are combined through reactions to form the final molecule during synthesis. The state, on the other hand, refers to the molecule’s 3D structure, i.e., the 3D coordinates of its atoms. The paper proposes CGFlow (Compositional Generative Flow), a method based on flow matching—a generative modeling technique—that jointly and interdependently generates a molecule’s composition and state. This approach is specialized for designing synthetically feasible ligands through the 3DSynthFlow model [1].
The process of molecule generation with 3DSynthFlow is illustrated in the figure below. In a single-generation process, fragments are added to the molecule’s 2D structure (i.e., its composition) at regular intervals. As fragments are added to the 2D structure, corresponding atoms are also incorporated into the 3D structure (i.e., the state). Unlike the 2D structure, the 3D structure is continuously refined at each step to achieve a more precise conformation. Once the generation process is complete, a fully refined molecule is produced in both 2D and 3D representations.
The authors demonstrated two key performance aspects of 3DSynthFlow: (1) optimizing activity for a single target and (2) generating ligands for diverse (new) targets using a single model. For (1), as shown in the table below, 3DSynthFlow generated ligands with more optimized docking scores compared to similar models.
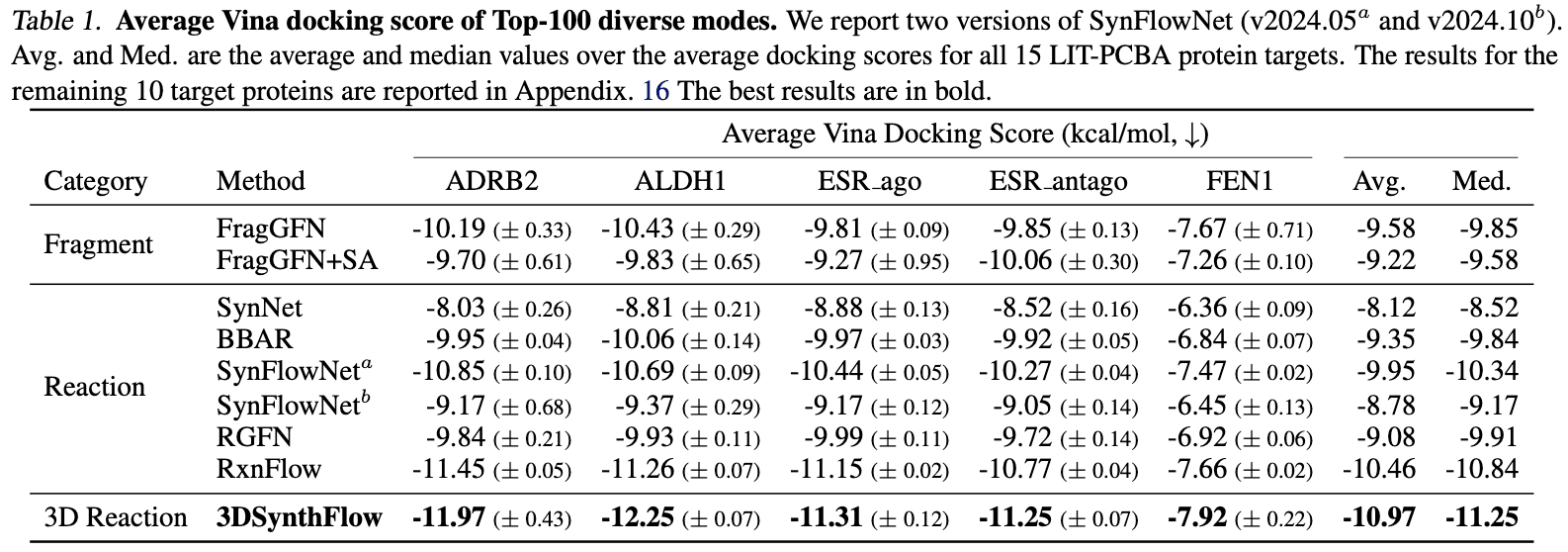
For (2), it achieved the best performance in both docking scores and synthetic feasibility across 100 diverse targets. Synthetic feasibility was evaluated using AiZynthFinder [Genheden et al. 2020].
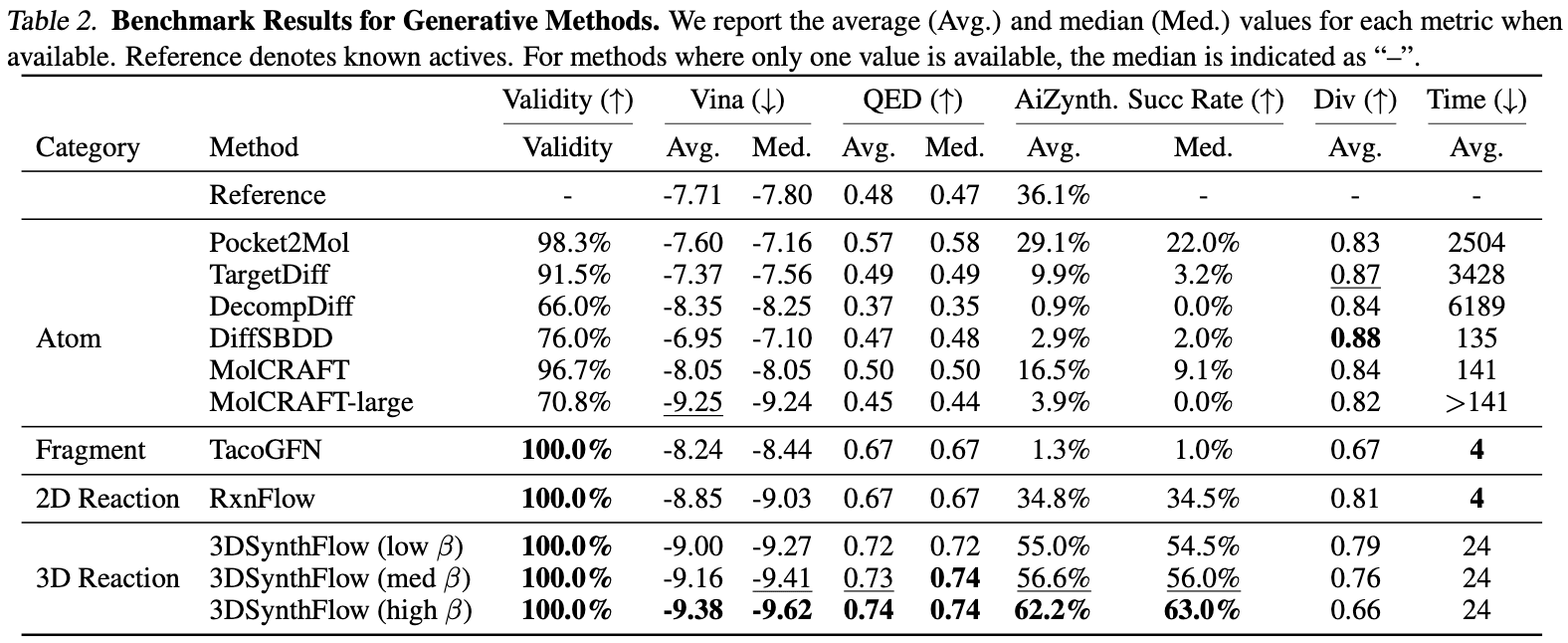
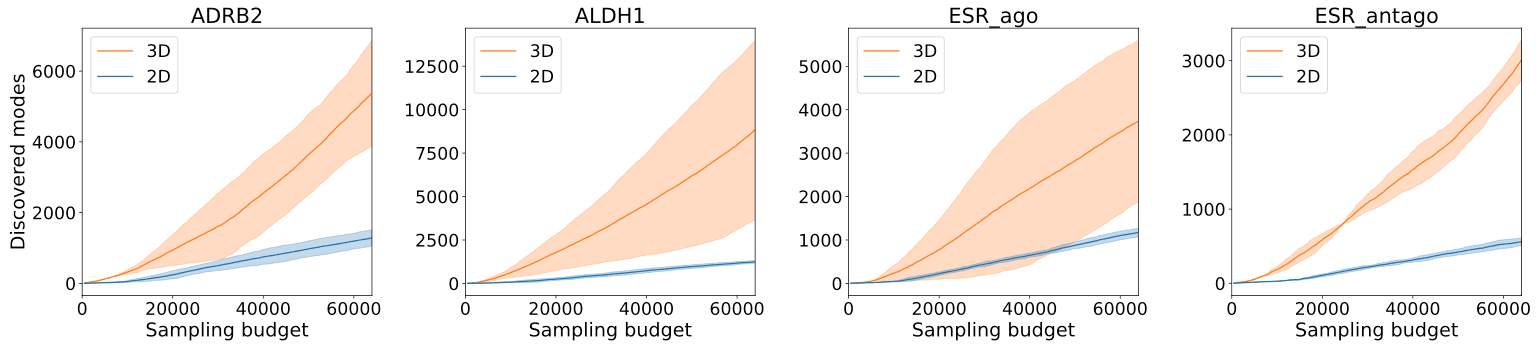
Additionally, compared to the authors’ previous model, which only handled 2D structures, 3DSynthFlow’s incorporation of 3D structure information enabled the discovery of more molecular structures per unit of time.
ICML 2025 Preview 2 | CDN: Target Discovery Through Causal Networks
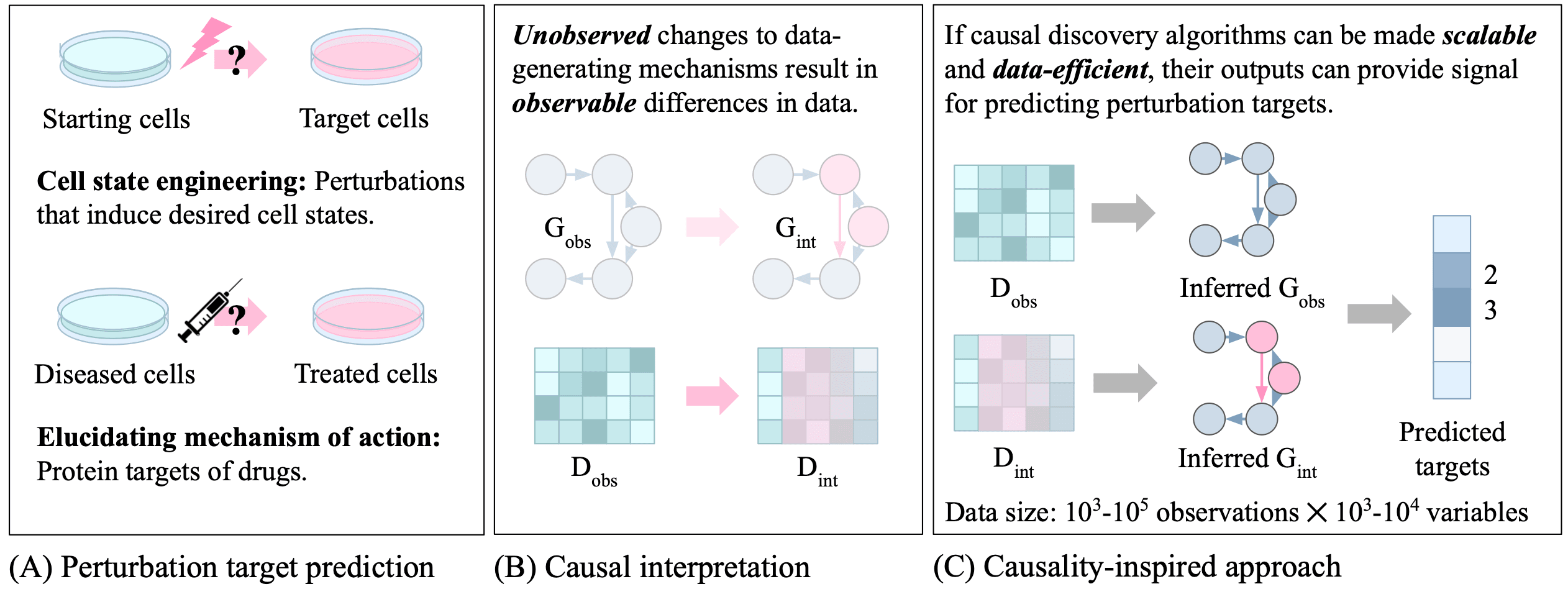
Target discovery is arguably the most critical initial step in drug development. Failing to accurately identify the target for treating a specific disease or the primary target of a drug’s action can render subsequent research ineffective or reveal the side effects of a marketed drug only years later.
The paper “Identifying biological perturbation targets through causal differential networks” by Menghua Wu, Umesh Padia, and colleagues introduces a deep learning model for predicting target genes responsible for specific perturbations (interventions). The proposed model, CDN (Causal Differential Networks), requires transcriptomic datasets from the same cell line before and after a perturbation. From each dataset, a causal graph is first estimated to represent (i.e., generate) the data. In these causal graphs, nodes represent genes, and edges represent relationships between genes. After estimating the pre-perturbation and post-perturbation causal graphs from the two datasets, the differences between the graphs are used to predict the genes targeted by the perturbation. In other words, CDN operates on the principle that changes in underlying causal relationships are responsible for the observed data changes before and after the perturbation.
CDN evaluated its target prediction performance on two transcriptomic datasets. First, it was tested on CRISPR-based Perturb-seq data [2], where, as shown in the table below, it demonstrated significantly superior prediction performance compared to baseline methods. The evaluation data was separated such that the gene perturbations differed from those in the training data. Additionally, CDN maintained its performance even when the data was further separated by cell line (referred to as “Unseen Cell Line” in the table below).
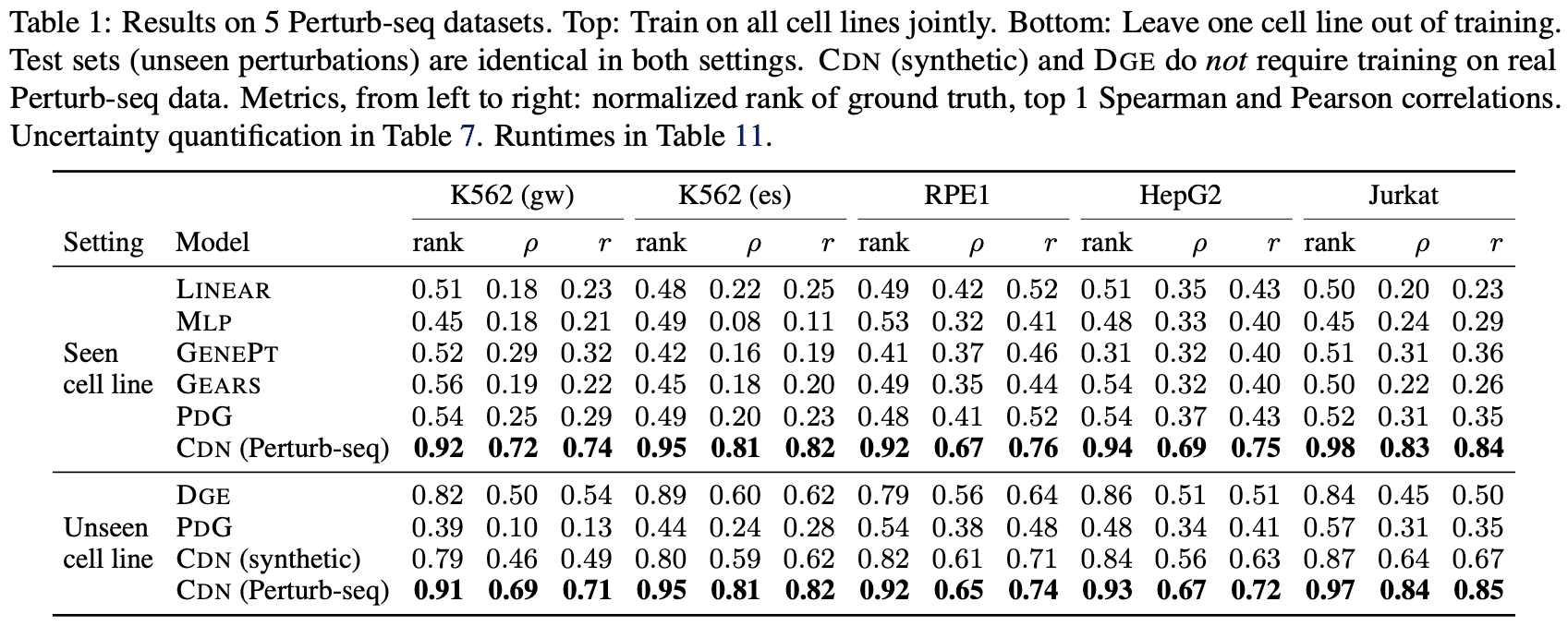
For the second evaluation, sci-Plex data [3], which involves gene expression changes induced by drugs, was used. Drug-induced perturbations affect genes much more indirectly than CRISPR, making it more challenging to identify underlying targets. In this scenario, CDN correctly identified the targets for 3 out of 6 drugs across two cell lines. However, the performance dropped significantly for some drugs (Infigratinib and Palbociclib), suggesting that discovering targets across diverse cell lines and drugs remains a highly challenging task.
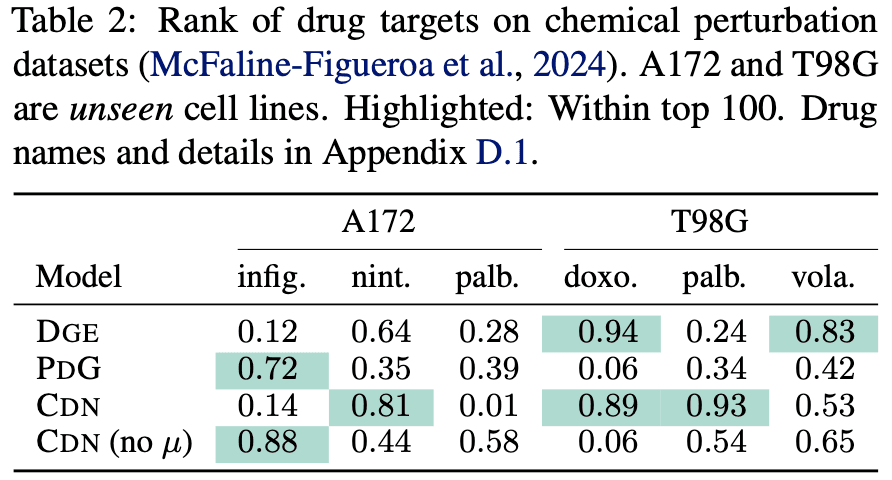
CDN has several advantages, including its independence from external biological data beyond input transcriptomic data and its significantly faster computational speed compared to competing models. However, it is somewhat disappointing that the paper does not analyze whether the causal graphs predicted as intermediate representations hold biological meaning or interpretability.
CML 2025 Preview 3 | Semi-Clipped & PEA: Connecting Morphological and Genetic Features of Cells
Two prominent data modalities for studying cellular mechanisms are transcriptomic data and microscopic image data. Transcriptomic data captures the types and quantities of genes expressed in cells, while image data represents the morphological characteristics of cells. By using both transcriptomic and image data from the same cells, could we learn generalized patterns that explain a wider range of cellular states and predict cellular properties more accurately?
In the paper “A Cross-Modal Knowledge Distillation & Data Augmentation Recipe for Improving Transcriptomics Representations through Morphological Features” by Ihab Bendidi, Yassir El Mesbahi, and others from Recursion, Valence Labs, École Normale Supérieure - PSL, the authors propose an answer to this question by adapting OpenAI’s well-known CLIP (Contrastive Language–Image Pre-training) method. Originally, CLIP was designed to train deep-learning models to connect images with their corresponding text descriptions. As shown in the figure below, its core principle involves embedding images and text in a shared space, where embeddings of related image-text pairs are trained to be similar, while unrelated pairs are trained to be dissimilar.

In this paper, the authors define microscopic cell images as the teacher modality and transcriptomics as the student modality, training the student to learn the teacher’s features using the CLIP mechanism. The key difference is that image embeddings are fixed, and only transcriptomic embeddings are optimized based on CLIP’s objective function. This approach is termed Semi-Clipped. Semi-clipping enables (1) predictions using only one modality (transcriptomics) after training, while (2) distilling knowledge from image data into transcriptomic data, improving prediction accuracy compared to training with transcriptomic data alone.
The paper also addresses the batch effect—a common issue in cellular experimental data where results vary due to environmental factors, timing, or experimenters—through a data augmentation strategy. The authors apply one of several predefined normalization methods randomly to transcriptomic data during training, while consistently applying a single normalization method to image data. The CLIP objective function is computed using the normalized transcriptomic and image embeddings, training the transcriptomic embedding network for knowledge distillation. This randomness not only mitigates batch effects but also augments the training data, addressing the scarcity of paired transcriptomic-image data. Since obtaining both data types from the same cells is challenging, their correlation is considered only up to factors like cell lines or perturbation conditions. This augmentation method is named PEA (perturbation embedding augmentation).
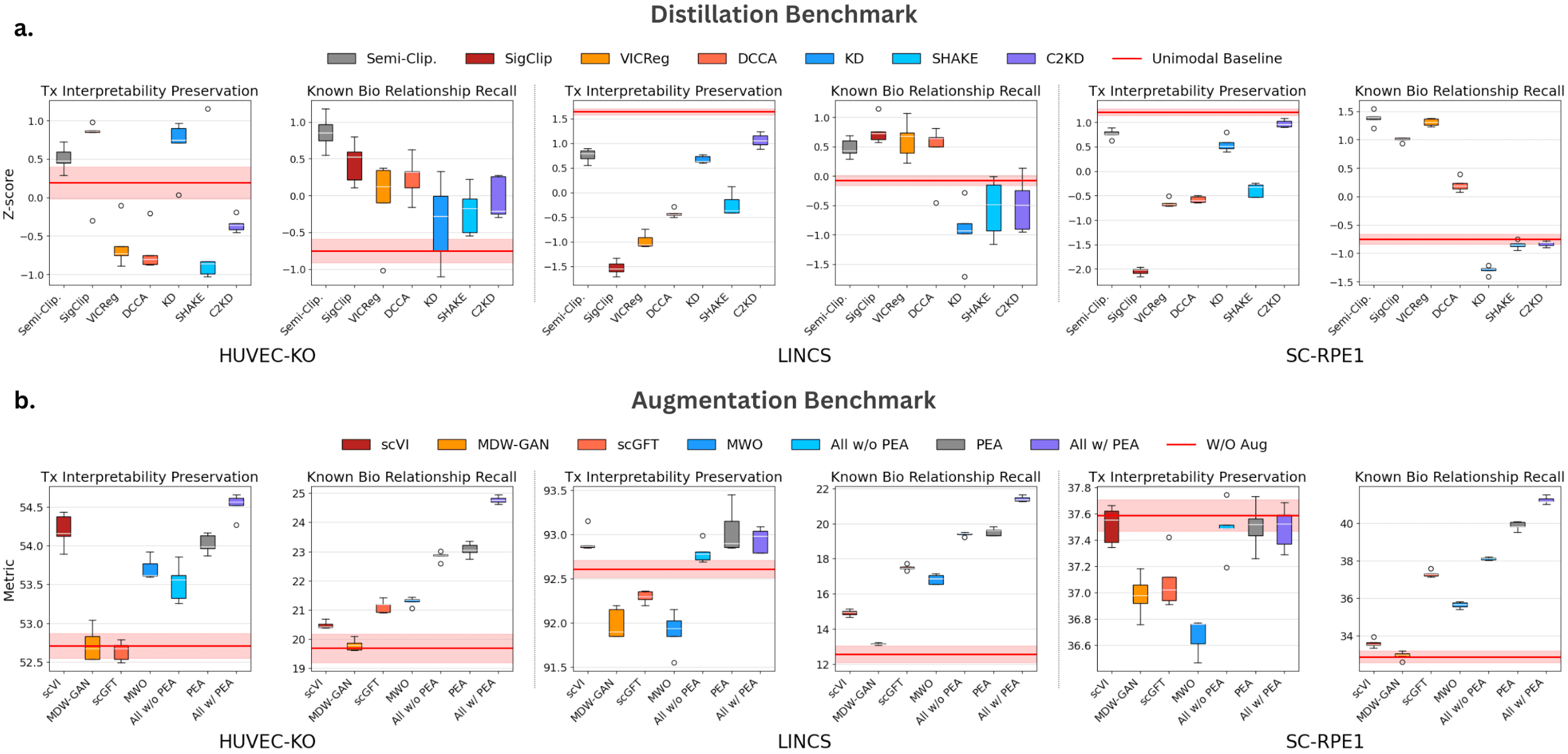
The proposed distillation and augmentation methods are evaluated on three out-of-distribution datasets. Two aspects are assessed for each method: (1) transcriptomic interpretability preservation, which measures how well the transformed transcriptomic embeddings retain the original transcriptomic data’s core information after distillation, and (2) known biological relationships retrieval, which evaluates how effectively the learned embeddings capture known gene relationships. As shown in the graph below, higher values indicate better performance and Semi-Clipped with PEA outperforms baseline methods in both aspects. The significance of this work lies in evaluating the method’s generalization across diverse variables, such as perturbation types (chemical vs. CRISPR), assay methods, and bulk vs. single-cell data.
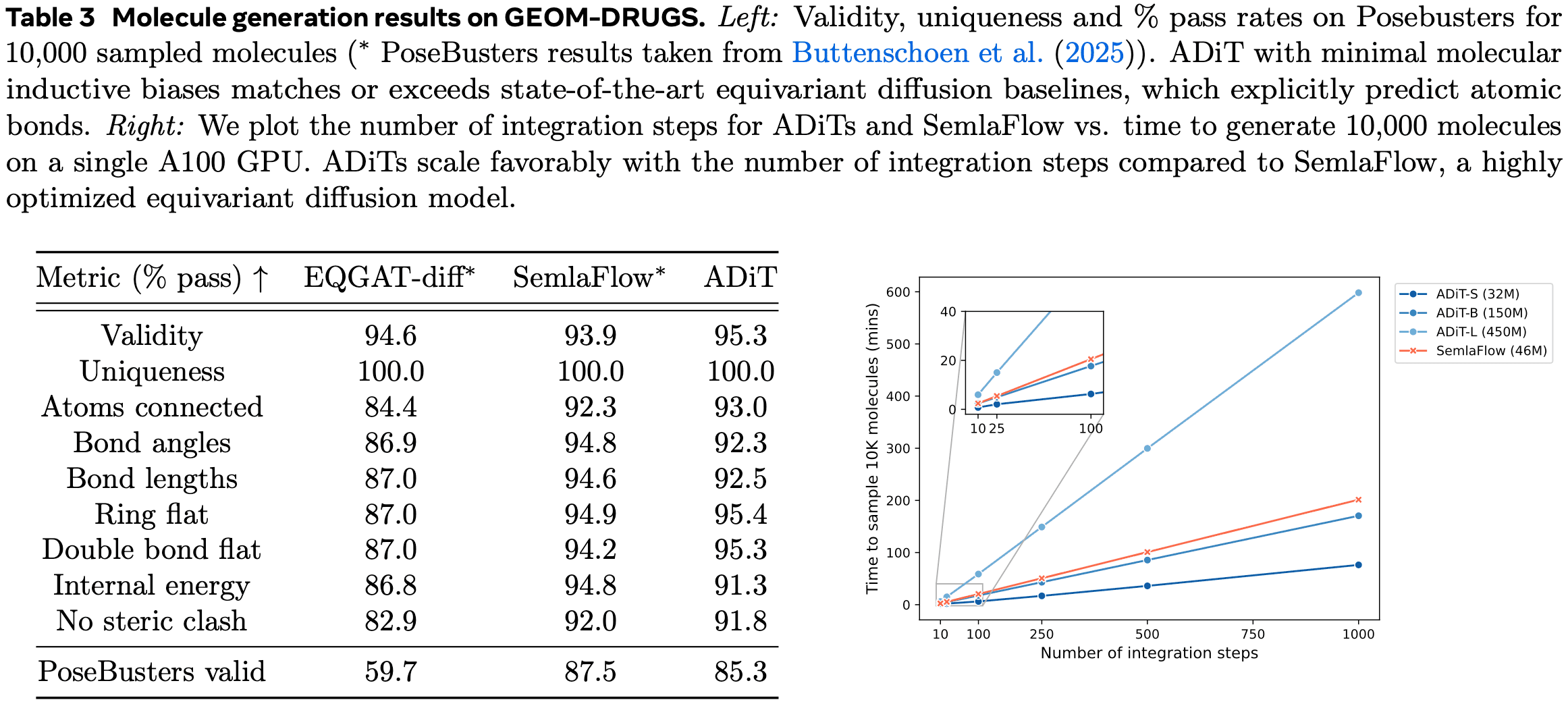
ICML 2025 Preview 4 | ADiT: A Foundation Model for Molecular and Material Systems
As generative AI for creating 3D molecular structures continues to advance, recent interest has shifted toward models capable of handling molecular systems across multiple scales, from small molecules to large macromolecules like proteins. In “All-atom Diffusion Transformers”, to be presented by Meta’s FAIR (Fundamental AI Research), a different approach is introduced, proposing a method to generate both crystalline and amorphous molecular systems within a single model.
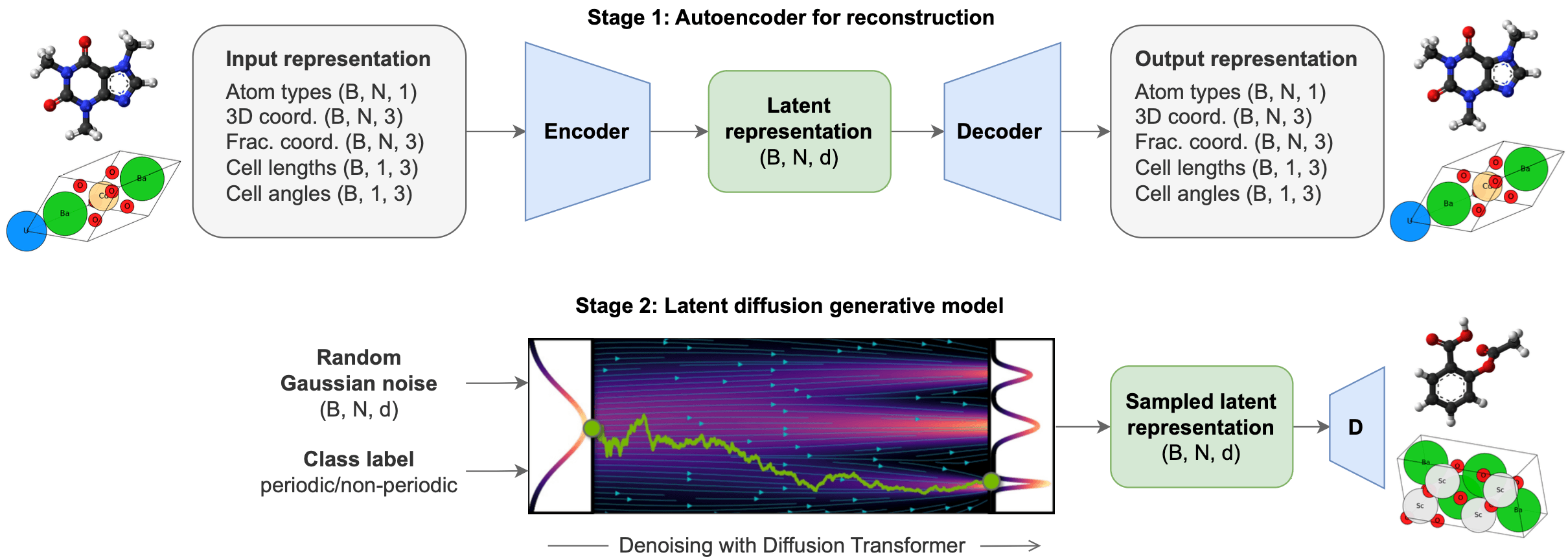
The framework of this paper, ADiT (all-atom diffusion transformer), first learns a latent space where both crystalline and amorphous structures are embedded using a Variational Autoencoder (VAE). The VAE’s decoder is trained to reconstruct 3D crystalline or amorphous molecular structures from these embeddings.
Next, a Diffusion Transformer (DiT) is employed to generate novel structures. ADiT applies the principles of diffusion models, already known for their excellent performance in 3D molecular structure generation, to the VAE’s latent space (latent diffusion). To generate new structures, an embedding is randomly sampled from a standard normal distribution and undergoes a denoising process via the DiT. The denoised embedding is then reconstructed into a real-space molecular structure by the VAE’s decoder.
The figure below illustrates the all-atom embedding and DiT process of ADiT as described above.
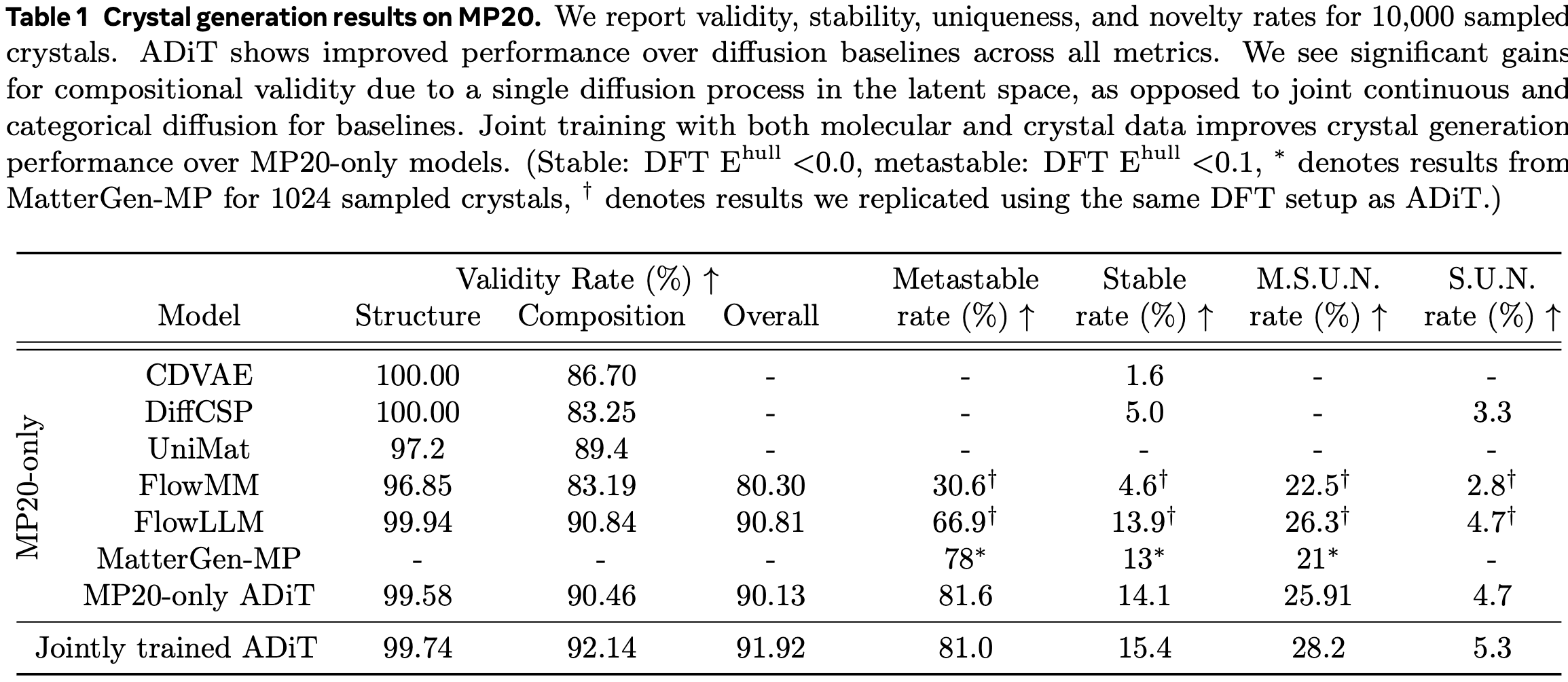
As ADiT highlights its ability to handle both crystalline and amorphous systems simultaneously as a key advantage, its generative performance was evaluated for each system. First, as shown in the table below, ADiT demonstrated state-of-the-art performance in generating crystalline structures. Additionally, it was observed that training on both crystalline and amorphous molecular data together improved performance, though the improvement appears to be relatively modest.
Next, the generative performance of amorphous molecular structures was evaluated. Another distinctive feature of ADiT is the omission of equivariance, which is commonly incorporated in 3D structure generation [4]. ADiT addresses this through data augmentation. In the table below, the baseline models EQGAT-diff and SemlaFlow both adopt structures that ensure equivariance. In contrast, ADiT, despite not guaranteeing equivariance, achieves superior or comparable performance with significantly lower computational costs. This allows ADiT to scale up model size easily and train on larger datasets.
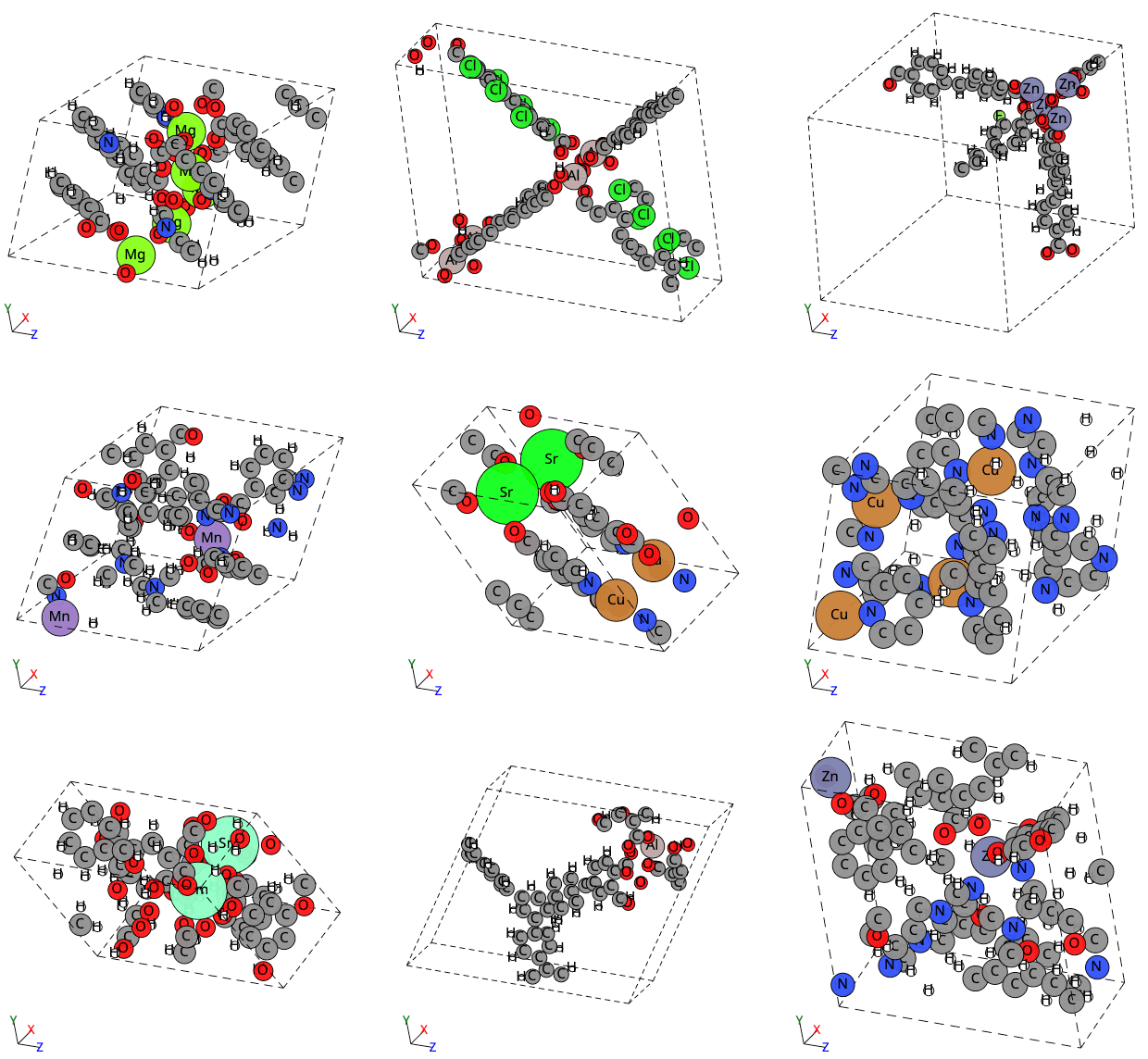
Finally, visual examples are provided to showcase ADiT’s generation of metal–organic frameworks (MOFs).
The most significant limitation of the ADiT paper is that the demonstrated systems are relatively small. The evaluated systems contain slightly over 100 atoms at most, whereas systems like proteins or nucleic acids consist of hundreds or thousands of atoms. The authors acknowledge this point and suggest that ADiT’s high scalability offers potential for addressing this issue.
Soon, perhaps a true foundation AI, like AlphaFold 3, capable of generating biomolecular systems across various scales as well as crystalline material structures simultaneously, will emerge. Whether such a unified AI model would inherently offer unique advantages is unclear, but it seems likely to aid in learning the fundamental physical principles underlying all atomic systems.
Stories Left Untold
Due to constraints, we could only cover individual papers as described above. However, as mentioned earlier, interest in AI4Science is rapidly growing, leading to the emergence of many intriguing studies. While papers applying large language models (LLMs) to problems like drug discovery continue to be actively published, they were omitted from this preview. LLM applications are particularly prevalent in the medical field compared to drug discovery. You can explore related papers by searching for terms like “medical” on the ICML 2025 paper listing page.
In the upcoming NeurIPS 2025 preview in the second half of the year, we’ll dive into even more exciting research.
[1] To be precise, CGFlow is introduced as a generalized method applicable to any target that can be represented with compositional stages and continuous variables, without specifying molecules.
[2] Dixit et al. 2016; Replogle et al. 2022; Nadig et al. 2025.
[3] Srivatsan et al. 2019; McFaline-Figueroa et al. 2024.
[4] A helpful resource for understanding the importance of molecular systems and equivariance [White 2021].