이번 AlphaFold 3 리뷰도 이전 포스트와 마찬가지로 전체 내부 구조와 흐름을 입력 데이터 구성 → 내부 업데이트 모듈 → 최종 3차원 구조 예측 알고리즘 순으로 알아보는 형식으로 진행하겠습니다.
AI
AlphaFold 3 리뷰 - Google DeepMind, 신약 개발의 새로운 패러다임을 제시하다
AlphaFold 3가 약 9개월간 베일에 싸여있다가 이번 논문을 통해 드디어 세상에 공개됐습니다. AlphaFold 2와는 어떤 점이 크게 달라지고, 발전했을까요? 이번 포스트에서는 AlphaFold 3에 대해 최대한 쉽고 간략하게 알려드립니다.

배성한 AI 연구원
2024.06.1420min read

AlphaFold 3, 세상에 그 모습을 드러내다
안녕하세요. 히츠의 AI 기술 연구 개발을 담당하고 AI 연구원 배성한이라고 합니다.
지난 5월 8일, AI 및 신약 개발 분야를 비롯한 과학계 전체를 흥분시킬 만한 소식이 전해졌습니다. 바로 구글 딥마인드(Google DeepMind)에서 차세대 AlphaFold, AlphaFold 3에 대한 논문을 발표한 것입니다. AlphaFold 3는 지난 2023년 8월에 딥마인드에서 그 등장을 처음 예고한 직후 단일 단백질뿐만 아니라 단백질-단백질, 단백질-핵산, 단백질-리간드에 이르기까지 거의 모든 유형의 단백질 기반 생체 분자 복합체의 3차원 구조를 정확히 예측할 수 있다는 점에서 많은 화제를 모으고 있습니다. 이 놀라운 기술이 약 9개월간 베일에 싸여 있다가 이번 논문을 통해 드디어 세상에 공개된 것입니다.
해당 논문은 발표된 지 한 달이 약간 안 된 현재 시점(2024.06.05) 기준으로 벌써 인용 수가 25회에 달할 정도이고, AlphaFold 3를 필두로 한 AI 기반 신약 개발 분야는 현시점 가장 주목받는 신기술 중 하나입니다. 따라서 저희 히츠 AI 연구팀과 같은 AI 연구자들은 물론이고 제약바이오 분야에 종사하시는 많은 분들께서 이번에 공개된 AlphaFold 3에 대해 관심이 많으실 거라고 생각되는데요. 하지만 AlphaFold 3는 현재 전 세계의 AI 기술 발전을 주도하고 있는 그룹 중 하나인 딥마인드의 작품답게 매우 복잡하고 정교하게 설계된 모델이라 AI 기술에 익숙하지 않으시면 이해가 어려울 수 있습니다. 이번 포스트에서는 여러분들의 이해를 돕고자 AlphaFold 3에 대해 최대한 쉽고 간략하게 리뷰하고자 합니다.
전체적으로 AlphaFold 3는 이전 버전인 AlphaFold 2를 기반으로 재설계된 모델이라고 볼 수 있을 만큼, 내부 구조나 알고리즘 측면에서 유사한 점이 많습니다. 따라서 이번 리뷰에서는 AlphaFold 3가 AlphaFold 2와 비교했을 때 어떤 부분이 달라졌는지를 중심으로 설명드리려고 합니다. AlphaFold 2에 대한 사전 지식이 없으신 분들은, 이를 다룬 이전 포스트를 먼저 참고하신 뒤 읽어보시는 것을 추천드립니다. 그럼 본격적으로 AlphaFold 3 리뷰를 시작하겠습니다.
AlphaFold 3, 무엇이 달라졌나
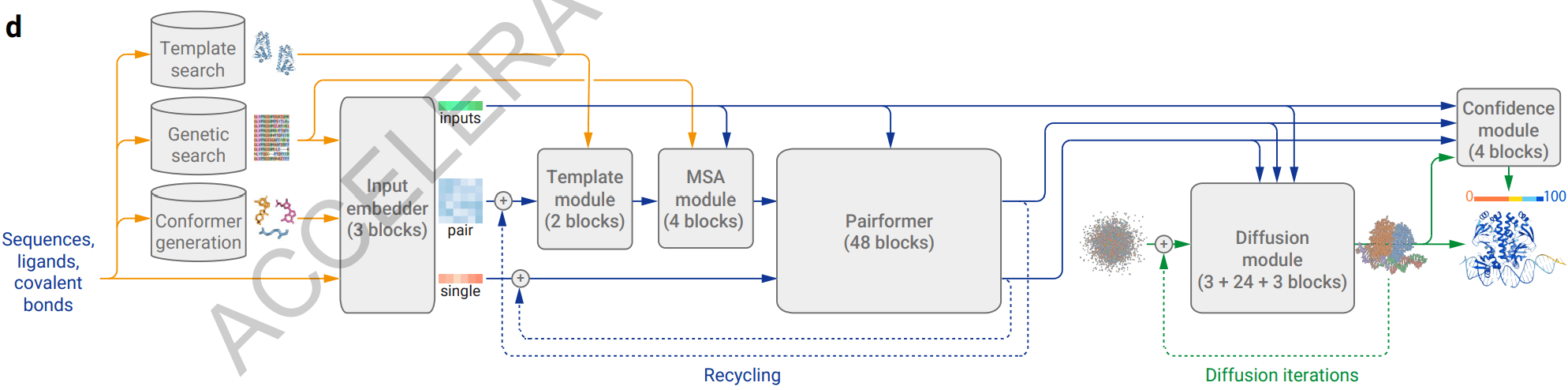
AlphaFold 3 변화점 - 1. 입력 데이터 구성 및 업데이트
먼저 입력 데이터부터 살펴보겠습니다.
이전 포스트에서 설명한 바와 같이, AlphaFold 2는 구조를 예측하고자 하는 단백질 서열에 대한 진화론적인 힌트를 주는 다중 서열 정렬(Multiple Sequence Alignment, MSA) 데이터와 구조적인 힌트를 제공하는 template 데이터를 기반으로 형성한 pair representation, 이 두 가지 데이터를 입력으로 받습니다. 이때 AlphaFold 2는 오직 단일 단백질에 대한 구조를 예측하는 모델이기 때문에, 이 두 데이터의 기본 단위는 단백질 서열의 구성 요소인 아미노산이 됩니다.
반면에 AlphaFold 3는 앞서 언급한 것처럼 예측하고자 하는 대상이 단백질뿐만 아니라 핵산과 리간드도 포함할 수 있기에 입력 데이터의 구성이 달라지기 때문에, 입력 서열을 각 구성 성분의 유형에 따라 서로 다른 입력 단위로 표현됩니다. 즉 입력 서열 내에 단백질은 아미노산, 핵산은 뉴클레오타이드, 그리고 리간드는 원자 단위로 표시하여 초기 입력 데이터를 구성합니다. 이렇게 구성된 입력 데이터는 Input Embedder에서 각 요소가 합쳐진 복합체 단위로 업데이트를 진행합니다.
그렇다면 서로 다른 단위를 가지는 입력 데이터를 어떻게 하나의 복합체로 통합한 뒤 업데이트할 수 있까요? 정답은 아미노산이나 뉴클레오타이드 단위들을 최소 입력 단위인 원자 단위로 쪼개어 연산을 수행하는 것입니다. 이렇게 원자 단위로 입력을 세분화하면 임의의 생체 분자 복합체가 입력으로 들어오더라도 원자라는 공통된 입력 단위를 토대로 전체 복합체를 표시할 수 있고 추후 하나의 일관된 연산을 적용할 수 있게 됩니다.
이때 reference conformer라는 추가 데이터가 사용되는데, 이것은 아미노산이나 뉴클레오타이드와 같은 분자 단위의 입력을 원자 단위로 쪼갤 때 필요한 각 분자의 원자 구성 및 구조 정보를 제공합니다. 이렇게 재구성된 입력 데이터는 Input Embedder 네트워크 내에서 원자 단위의 연산을 통해 업데이트됩니다. 이때 각 원자마다 상관관계(attention)를 고려하는 연산을 수행하기 때문에 이 연산을 AtomAttention이라고 부릅니다.
AtomAttention 연산 결과값은 다시 원래 입력 단위로 재구성되어 아래 figure에서 single로 표현되는 업데이트된 입력 서열을 형성합니다. 이는 AlphaFold 3가 AlphaFold 2와 마찬가지로 MSA 및 template 데이터 사용하여 추가적인 업데이트를 진행하기 때문입니다. MSA 와 template 데이터는 아미노산 혹은 뉴클레오타이드 단위로 표현되기 때문에 AtomAttention 연산 결과 내에 원자 단위로 쪼개진 아미노산/뉴클레오타이드 표현을 다시 분자 단위 표현으로 합쳐주는 것이죠. 그리고 업데이트된 입력 서열에 외적 (outer product) 연산을 적용하여 pair representation을 형성한 뒤, 이를 순차적으로 Template module 과 MSA module을 통해 각각 template, MSA 데이터의 정보와 결합하여 업데이트 해줍니다.
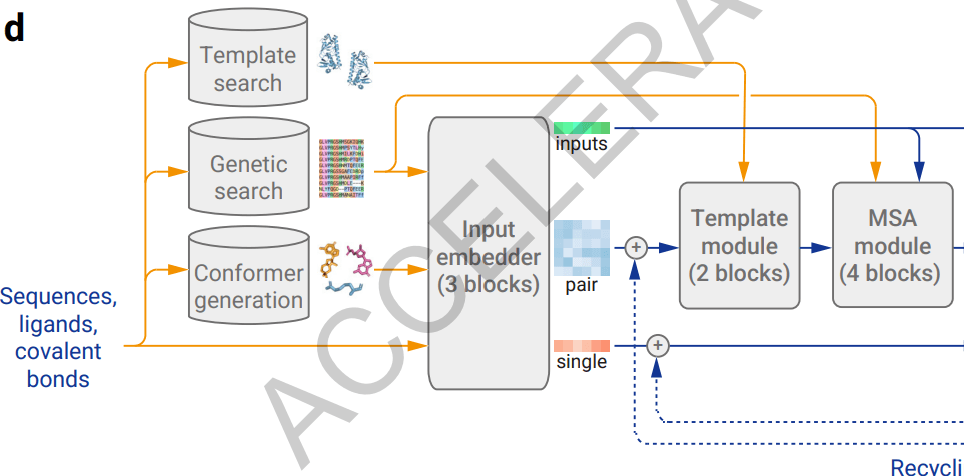
AlphaFold 3의 입력 데이터 구성과 업데이트 과정은 아래와 같이 요약할 수 있습니다.
- 입력 : 입력 서열
- 단백질은 아미노산, 핵산은 뉴클레오타이드, 그리고 리간드는 원자 단위로 입력 단위를 구성하여 초기 입력 데이터를 구성
- 입력 데이터를 reference conformer 데이터를 활용하여
Input Embedder내에서 원자 단위로 쪼갠 뒤 원자 단위로 상관관계를 고려하며 업데이트하는AtomAttention연산을 수행 AtomAttention결과 업데이트된 single representation을 기반으로 pair를 형성한 뒤,Template & MSA module내에서 template & MSA 데이터의 정보를 결합하여 업데이트
- 출력 : 업데이트 된 single 및 pair representation
AlphaFold 3 변화점 - 2. 내부 모듈의 변화 : Evoformer에서 Pairformer로
AlphaFold 2에서는 MSA 데이터와 pair representation 데이터를 Evoformer라는 네트워크를 통해 업데이트하였습니다. Evoformer는 attention을 활용해, MSA 내에서는 서로 다른 단백질 서열 간의 진화론적 상관관계를, pair representation 내에서는 입력 단백질 서열 내의 아미노산 간의 상관관계를 고려하며 각 데이터를 업데이트했습니다. 추가로 Evoformer는 연산 도중 MSA와 pair representation 사이에 서로 정보를 한번씩 교환하는 과정이 존재하며, 두 입력 데이터가 서로의 정보를 반영하면서 업데이트되도록 유도하였습니다.
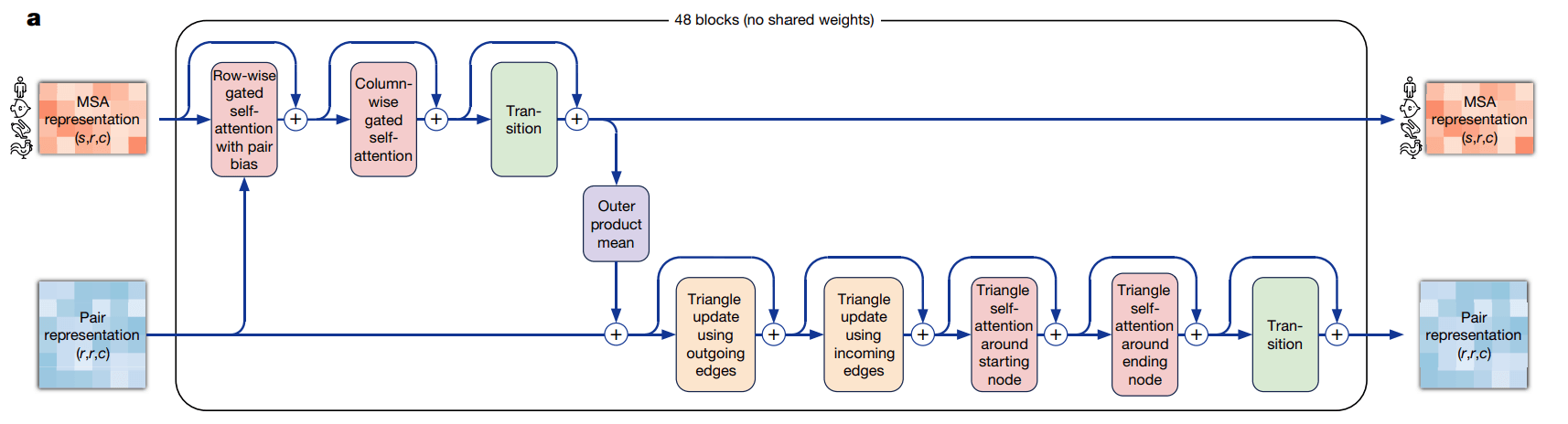
AlphaFold 3에서는 Evoformer와 유사한 Pairformer네트워크를 사용합니다. Pairformer와 Evoformer간의 가장 큰 차이점은 바로 입력으로 받는 데이터의 종류와 내부 연산입니다. Evoformer가 입력 서열과 진화론적으로 유사한 다른 단백질 서열까지 포함된 MSA 데이터를 입력으로 받는 대신에, Pairformer는 앞선 Input Embedder에서 업데이트된 입력 서열(single representation)을 받습니다. 따라서 Evoformer와 달리 MSA 내부에서 서열 간 attention 연산과 MSA 와 pair representation 간의 정보 교환 연산이 각각 업데이트된 서열(single representation) 내의 요소들끼리의 attention 연산과 pair에서 single representation으로의 하나의 정보 교환 연산으로 간소화됩니다. 이때 Pairformer에서 빠진 MSA와 pair representation간의 정보 교환 연산은 앞서 언급한 MSA module로 대체합니다.
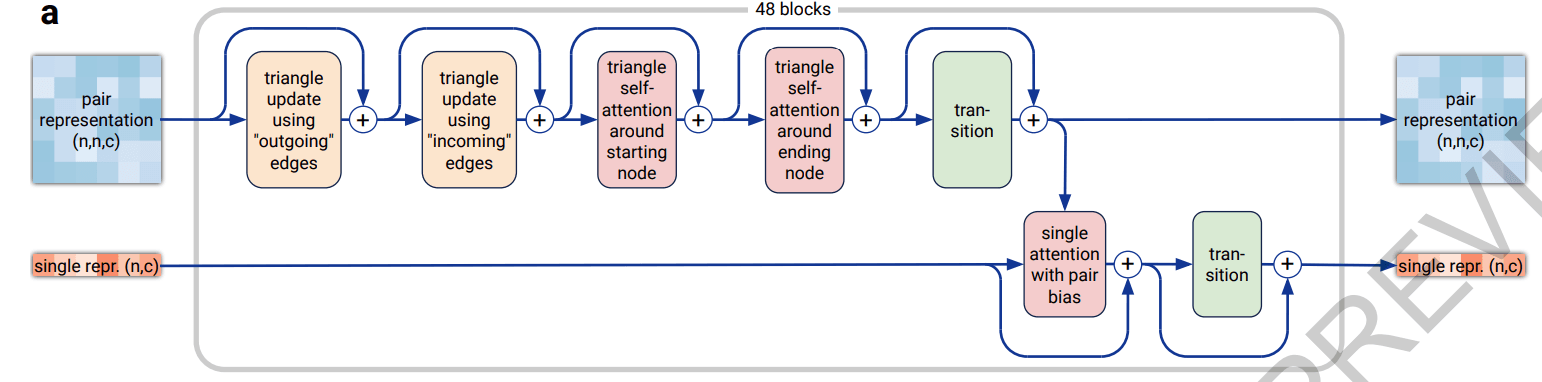
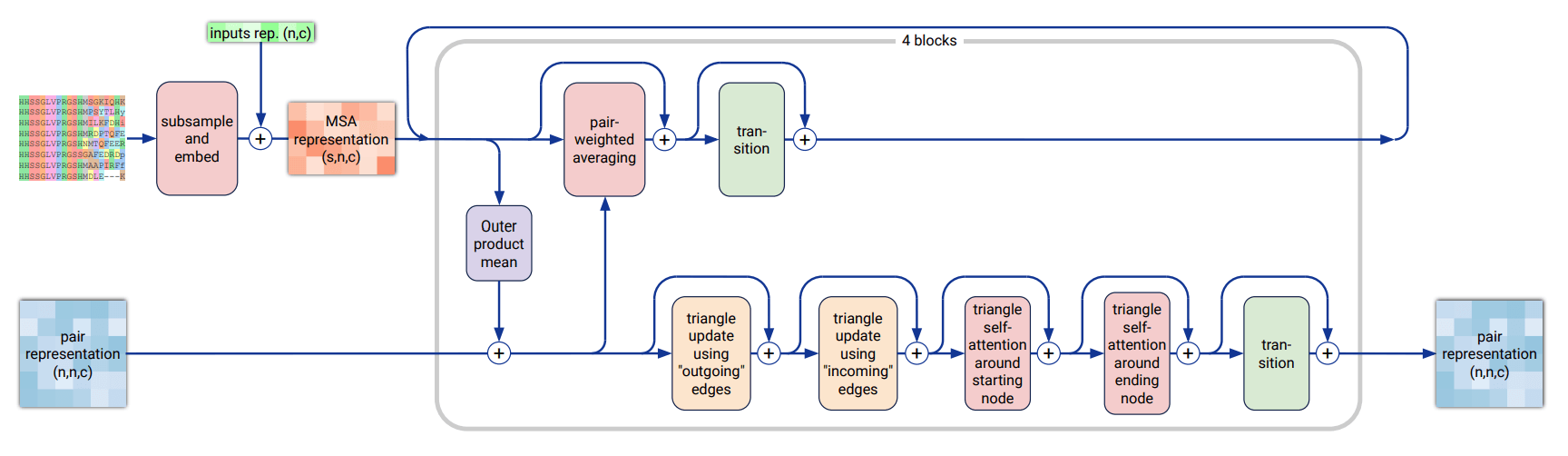
Pairformer내의 데이터 흐름과 연산을 요약하자면 아래와 같습니다.
- 입력 :
Input Embedder에서 업데이트한 single & pair representation
- Pair representation 내에 요소들끼리의 attention을 통한 연산을 통해 업데이트
- 업데이트된 pair representation의 정보를 single로 전달
- pair의 정보를 받은 single representation을 업데이트
- 출력 : 업데이트된 single & pair representation
정리하자면, Pairformer는 Evoformer를 토대로 입력으로 받는 데이터의 크기(여러 서열로 이루어진 MSA 데이터에서 입력 서열로만 이루어진 single representation으로)와 내부 연산(MSA ↔ pair 정보 교환이 pair → single로)이 간소화된 네트워크라고 할 수 있습니다.
Pairformer와 Evoformer는 각각 AlphaFold 3과 AlphaFold 2의 내부 구조에서 가장 큰 비중을 차지하는 네트워크인데요. 간소화된 Pairformer를 통해 AlphaFold 3는 AlphaFold 2에 비해 전체 연산에 쓰이는 계산 자원과 시간을 줄일 수 있습니다.
AlphaFold 3 변화점 - 3. 새로운 구조 예측 네트워크, Diffusion
AlphaFold 2는 Evoformer에서 업데이트된 MSA & pair representation을 기반으로 Structure module 을 통해 최종적으로 단백질의 구조를 예측했습니다. Structure module은 먼저 입력으로 받은 MSA & pair representation으로부터 아미노산들의 유클리디언 변환(Euclidean Transformation) 행렬을 예측하여 backbone의 위치를 구한 뒤, 각 아미노산 마다 뒤틀림각을 예측하여 아미노산 내의 개별 원자들의 좌표를 계산하는 방식으로 단백질 내 전체 원자들의 3차원 좌표를 예측합니다. 그리고 현재까지 예측한 단백질 구조 정보를 다시 Structure module의 입력으로 넣는 과정을 반복함으로써, 단백질 내 원자들이 원점에서 점진적으로 실제 좌표로 이동하도록 유도합니다.
AlphaFold 3는 생체 분자의 구조를 예측하기 위해, 이미지 생성에 널리 쓰이는 AI 모델 중 하나인 Diffusion을 사용했습니다. Diffusion이란 원본 데이터(이미지)에 점진적으로 노이즈를 추가한 뒤, 그것을 제거하는 과정을 네트워크를 통해 학습시켜, 최종적으로 노이즈 상태에서 학습한 데이터와 유사한 새로운 데이터를 생성할 수 있는 생성형 AI 모델입니다.
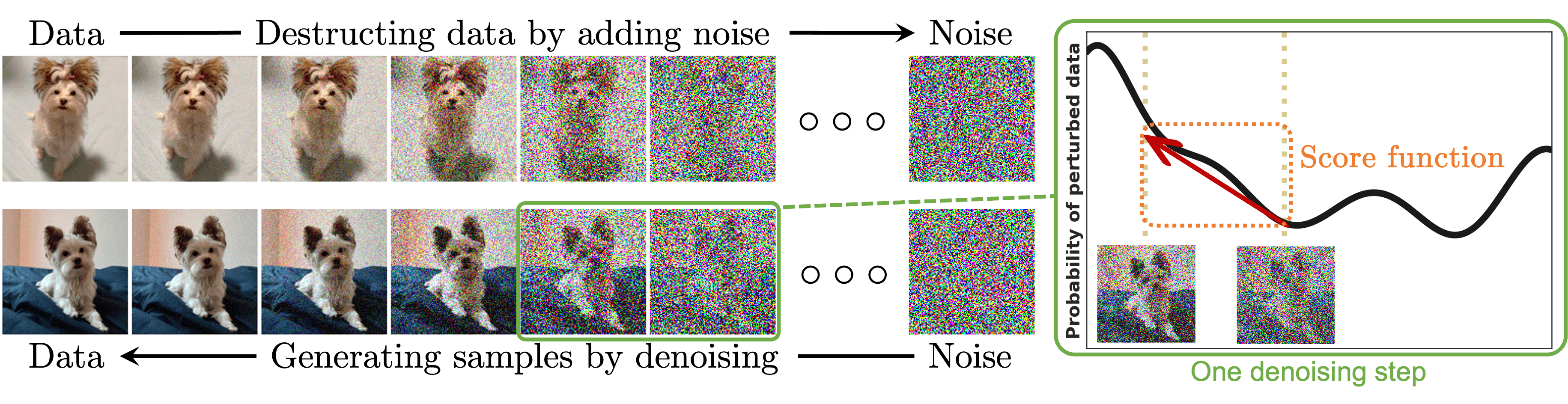
AlphaFold 3에서는 이 Diffusion을 기반으로 노이즈에서 생체 분자 내의 원자들의 3차원 좌표를 생성하는 Diffusion module로 Structure module을 대체했습니다. 먼저 Pairformer의 출력인 업데이트된 single & pair representation을 입력으로 받아서 Diffusion Conditioning 연산을 수행하여 각 원자의 3차원 내 공간적 조건을 계산합니다. 이때 single 및 pair representation에서 단백질과 핵산은 각각 아미노산과 뉴클레오타이드 단위로 표현되므로, 각 원자에 조건을 할당하려면 Input Embedder처럼 데이터 내 입력 단위를 다 원자 단위로 쪼개주는 과정이 포함됩니다. 이렇게 계산된 공간적 조건은 노이즈가 포함된 원자들의 3차원 좌표 정보와 결합되어, 노이즈가 추가되기 전의 올바른 좌표를 예측하는 데 사용됩니다.
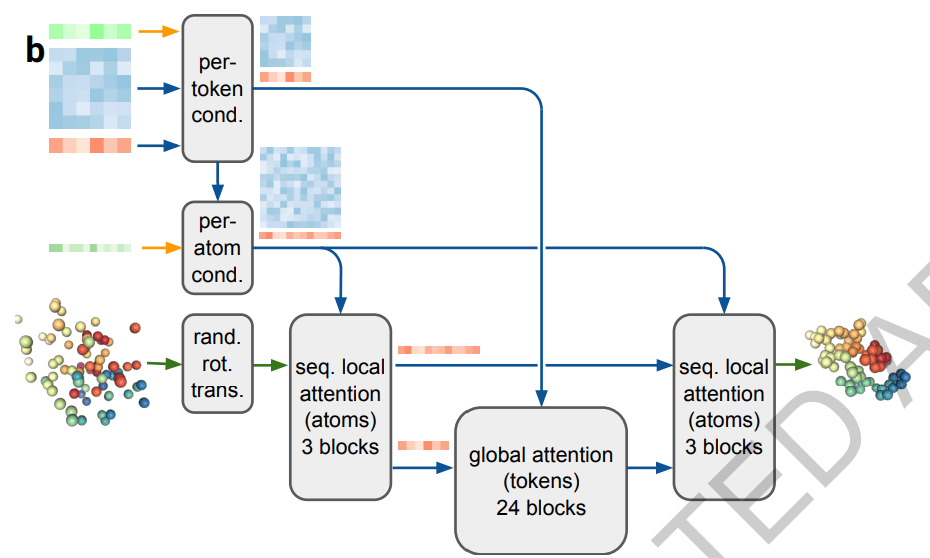
Diffusion module이 최종적으로 원자들의 3차원 좌표를 예측하는 과정은 아래와 같습니다.
- 입력 :
Pairformer에서 업데이트한 single & pair representation, 노이즈가 추가된 원자들의 3차원 좌표
- 업데이트된 single & pair representation을 원자 단위로 세분화한 뒤 각 원자마다 3차원 공간적 조건을 담도록 업데이트
- 업데이트 각 원자별 공간적 조건과 노이즈가 추가된 원자들의 3차원 좌표를 결합하여 노이즈가 제거된 원본 좌푯값을 예측
- 출력 : 노이즈가 제거된 원자들의 3차원 좌표
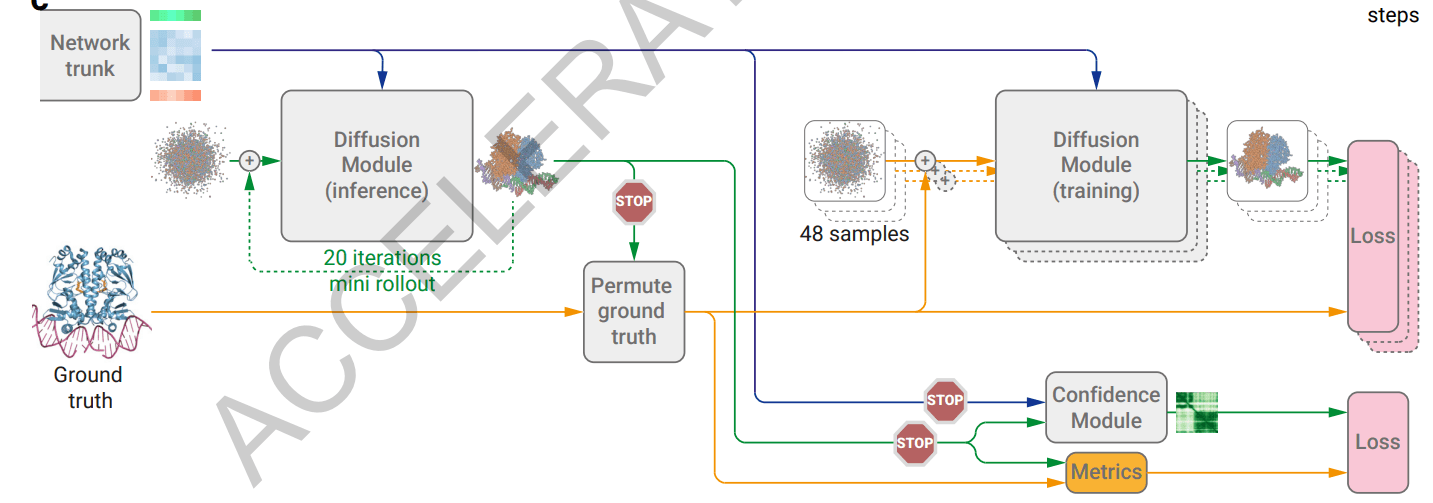
AlphaFold 3는 학습 시 생체 분자의 실제 3차원 구조 (Ground truth) 하나당 여러 개의 노이즈가 추가된 샘플을 생성하고, 각 샘플을 Diffusion module의 입력으로 넣어줘서 Ground truth의 3차원 좌표를 예측하도록 합니다. 즉, Diffusion module은 학습 과정에서는 노이즈를 한 번만 제거하는 single step of the diffusion을 학습하는 것이지요. 그러나 추론 과정에서는 single step of the diffusion을 열거하여 각 step마다 나온 노이즈 제거 결과를 다시 Diffusion module의 입력으로 넣어 주는 과정을 반복합니다. 이것은 AlphaFold 2에서 Structure module이 전 구조 예측값을 입력으로 받아 점진적으로 개선해 나가던 과정과 유사하며, 논문에서는 이를 mini-rollout이라고 부릅니다. 이러한 mini-rollout 과정을 통해 Diffusion module은 완전한 노이즈 상태에서 점진적으로 원자들의 3차원 좌표를 복원해낼 수 있게 됩니다.
AlphaFold 3의 등장이 우리에게 시사하는 점
지금까지 AlphaFold 3의 내부 구조 및 알고리즘에 대해 알아봤습니다. AlphaFold 3는 AlphaFold 2와 비교했을 때 크게 아래 3가지 요소들에서 특징적임을 알 수 있습니다.
- 임의의 생체 분자 복합체를 입력으로 받을 수 있게 입력 서열의 구성과 업데이트 방식 변화
Evoformer보다 간소화된Pairformer를 통해 singe & pair representation을 업데이트- 원자들의 3차원 좌표를 예측하기 위해서 생성형 AI 기술인
Diffusion을 접목
이 외에도 AlphaFold 3에는 하나의 포스트로는 모 다루기 어려울 만큼 많은 변화와 신기술이 녹아 있습니다. Supplementary Information을 살펴보면, AlphaFold 3를 설계하기 위해 저자들이 얼마나 깊이 고민하고 방대한 연구를 진행했는지를 여실히 느낄 수 있습니다. 그 결과 탄생한 AlphaFold 3는 단일 AI 모델로서 핵산과 리간드를 포함한 다양한 단백질 기반 생체 분자 복합체의 구조를 정확히 예측하며, 오늘날 AI 기반 신약 개발 분야의 게임 체인저로 자리매김하고 있습니다.
이번에 공개된 AlphaFold 3가 굉장히 놀랍고 혁신적인 기술임에는 틀림없지만, 여전히 한계는 존재합니다. 저희 AI 연구팀 황상연 팀장님이 AlphaFold-latest라는 제목으로 AlphaFold 3의 등장을 소개한 지난 포스트에서 언급하셨듯이, 현재 AlphaFold 3가 보여주는 구조 예측 성능은 지금까지의 State-of-the-Art(SOTA) 수준일 뿐, 절대적인 관점에서는 여전히 만점과는 거리가 있습니다. 또한 논문 내에서도 단백질-리간드 구조 예측에서 chirality가 제대로 반영되지 않거나 atom clashing이 발생하는 문제, 그리고 Diffusion을 포함한 생성형 AI 모델에서 자주 언급되는 고질적인 문제인 hallucination으로 인해 왜곡된 구조가 예측되는 등의 한계가 지적되었습니다. 즉, 현재 AlphaFold 3는 여전히 개선의 여지가 크며, 이를 시작으로 더 큰 진보가 이어질 것으로 기대됩니다.
AI 신약 개발이라는 기술적 격변의 시기에 대응하기 위해 전 세계적으로도 분주히 움직이고 있습니다. 실제로 이번 AlphaFold 3 논문이 코드가 첨부되지 않은 불완전한 오픈소스임이 드러나자 많은 연구자들 이에 대해 문의하였고, 해당 논문의 저널인 Nature에서 이례적으로 해명 기고를 낼 정도로 이번 AlphaFold 3의 등장과 AI 신약 개발이라는 혁신은 학계의 뜨거운 관심을 받고 있습니다. 이제 이 혁신이 실제 산업 현장에 반영되는 날도 머지않아 보입니다. 빠르게 변화하는 기술적 흐름에 압도되어 AI 신약 개발이 미지의 우주처럼 느껴지시는 분도 있으실 겁니다. 하지만 걱정하지 마시길. 국내외에서 인정받는 기술력을 보유한 AI 신약 개발 플랫폼인 히츠의 하이퍼랩과 함께라면 AI 신약 개발은 무궁무진한 가능성을 담은 기회의 장이 될 것입니다. 저희와 함께 AI 신약 개발이라는 미지의 영역을 마음껏 탐사해 보시죠.
AI 신약 개발이라는 기술적 격변의 시기에 전 세계는 분주히 움직이고 있습니다. 실제로 이번 AlphaFold 3 논문이 코드 없이 공개된 불완전한 오픈소스로 드러나자, 많은 연구자들이 이에 대해 문제를 제기했고, 해당 논문의 저널인 Nature에서는 이례적으로 해명 기고를 게재하기도 했습니다. 이는 AlphaFold 3의 등장이 AI 신약 개발이라는 혁신과 함께 학계에서 얼마나 큰 관심을 받고 있는지를 잘 보여줍니다. 이제 이 혁신이 실제 산업 현장에 반영되는 미래도 머지않았습니다. 빠르게 변화하는 기술 흐름에 압도되어 AI 신약 개발이 마치 미지의 우주처럼 느껴지실 수도 있습니다. 그러나 걱정하지 않으셔도 됩니다. 국내외에서 기술력을 인정받은 AI 신약 개발 플랫폼, 히츠의 하이퍼랩(HyperLab)과 함께라면, AI 신약 개발은 단순히 낯선 기술이 아니라 무궁무진한 가능성을 품은 기회의 장이 될 것입니다.
