ICLR 2025 Preview


What is ICLR?
Hi, I'm Seonghan Bae, a researcher from HITS AI Research Team 1. This April, the 2025 ICLR—an unmissable event for AI researchers—will take place at the Singapore Expo. ICLR (International Conference on Learning Representations) is an international AI conference established to enhance transparency through the OpenReview platform and to foster active discussions among AI researchers. It was first held in 2013 by deep learning pioneers such as Yoshua Bengio and Yann LeCun. Each year, ICLR showcases groundbreaking research in machine learning and AI, making it one of the most closely followed conferences by AI professionals worldwide—alongside ICML and NeurIPS, as noted in our previous post.
As AI-driven drug discovery continues to gain significant attention, the 2025 ICLR will showcase a diverse range of related research. This post provides a brief introduction to some of the most remarkable studies.
ICLR 2025 Preview: Generative Flows on Synthetic Pathway for Drug Design
Molecular generation models have recently become one of the most notable technologies in structure-based drug design (SBDD). By applying generative models—widely successful in image and natural language processing—to drug design, AI models can generate drug molecules (ligands) that are expected to effectively bind to specific target proteins.
This AI-driven approach to drug molecule generation is being hailed as a breakthrough technology, with the potential to replace traditional virtual screening methods that require substantial computational resources and time. However, most existing molecular generation models overlook 'synthetic feasibility,' making it challenging to synthesize proposed compounds, even when they are expected to have favorable properties.
The paper we are introducing today is titled Generative Flows on Synthetic Pathway for Drug Design. It presents an AI model called RxnFlow, which generates ligands with high synthetic feasibility by sequentially assembling molecules using predefined molecular building blocks and chemical reaction templates.
Key Methodology Overview
-
Sequential Molecular Generation Process
RxnFlow adopts the GFlowNets architecture to generate molecules in a sequential and learnable manner. GFlowNets, introduced in the 2021 paper GFlowNet Foundations, are an AI technique that learns the flow\(F(s\to s')\) from an initial state \(s_0\) to a terminal state \(s_f\). In this process, the total flow entering a state equals the total flow exiting that state, which is proportional to the probability of reaching that state. Furthermore, the flow reaching the terminal state is proportional to the reward \(R(sf)\), representing the desired outcome.
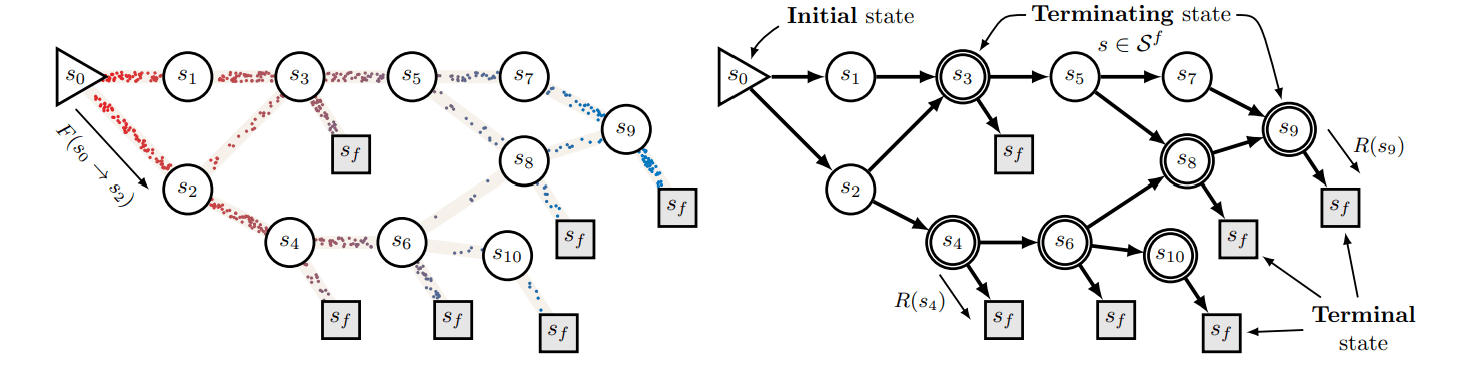
Figure 1. Schematic of GFlowNets [Source] BENGIO, Yoshua, et al. Gflownet foundations. The Journal of Machine Learning Research, 2023, 24.1: 10006-10060. In RxnFlow for ligand generation, the initial state consists of molecular building blocks, the terminal state represents the generated ligand, and the flow defines the synthetic pathway for assembling these building blocks into a complete molecule. Because this synthetic pathway is defined using actual building blocks and feasible reaction templates, the flow ensures high synthetic feasibility.
Thus, by defining the desired properties of a ligand for a specific protein—such as binding affinity to a target protein—as the reward, RxnFlow can generate diverse candidate ligands that meet these criteria while maintaining high synthetic feasibility.
- Action Space Sub-sampling
In RxnFlow, the action space is defined by combining 1.2 million building blocks with 71 reaction templates, resulting in synthesizable molecules. However, this space remains too large to be fully explored during training. To address this, RxnFlow applies an action space sub-sampling method for more efficient learning.
As shown in Figure 2 below, RxnFlow first embeds the building blocks and reaction templates using a neural network, then effectively reduces the action space through importance-based sub-sampling. Throughout training, actions are chosen within the reduced action space to transition from the current state to the next, ultimately guiding the model to maximize rewards at the terminal state. To account for the limited scope of the sub-sampled action space, weights are applied during this process.
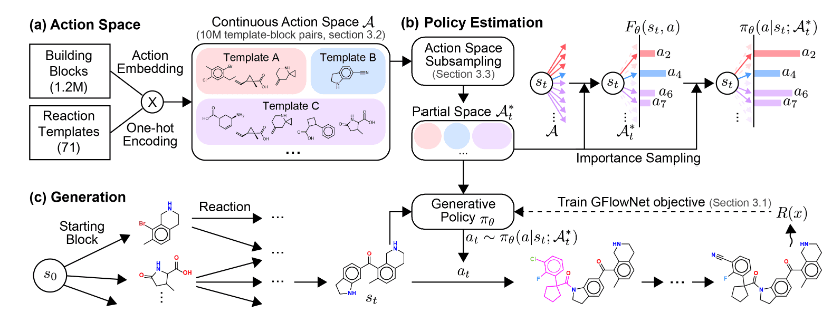
Figure 2. RxnFlow Training Process via Action Space Sub-sampling [Source] SEO, Seonghwan, et al. Generative flows on synthetic pathway for drug design. arXiv preprint arXiv:2410.04542, 2024. -
Non-Hierarchical MDP
In conventional synthesis-based molecular generation models, synthetic pathways are selected by first estimating the flow for the reaction template. Then, a hierarchical Markov decision process (MDP) is followed, where the flow progresses through building blocks based on the chosen reaction template. The authors of RxnFlow highlight a limitation of this approach, as shown in Figure 3(a). Once a reaction template is selected, only a limited set of building blocks can be used, which ultimately leads to suboptimal flows.
To overcome this limitation, RxnFlow introduces a non-hierarchical MDP approach, as illustrated in Figure 3(b). This method selects both the reaction template and the building blocks simultaneously, allowing for the stable learning of more diverse ligand generation flows.
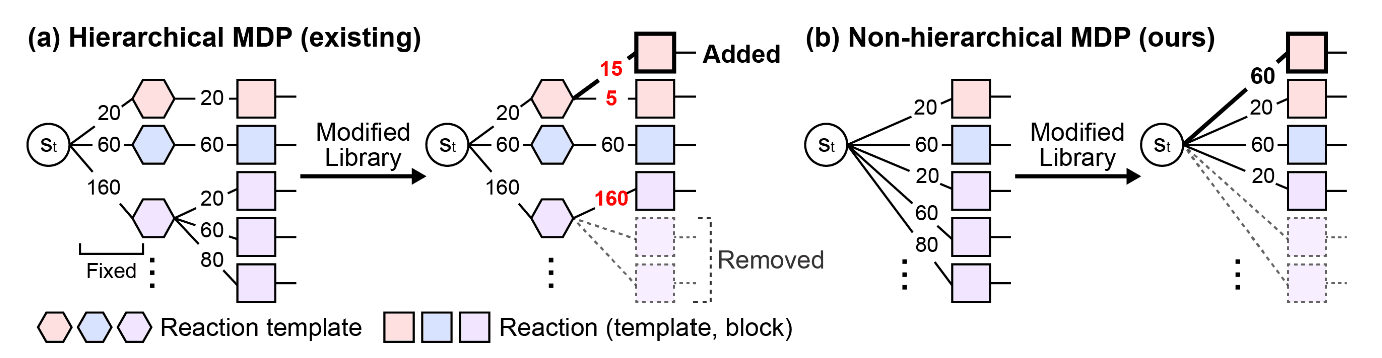
Figure 3. Non-Hierarchical MDP as Presented in RxnFlow [Source] SEO, Seonghwan, et al. Generative flows on synthetic pathway for drug design. arXiv preprint arXiv:2410.04542, 2024.
Results
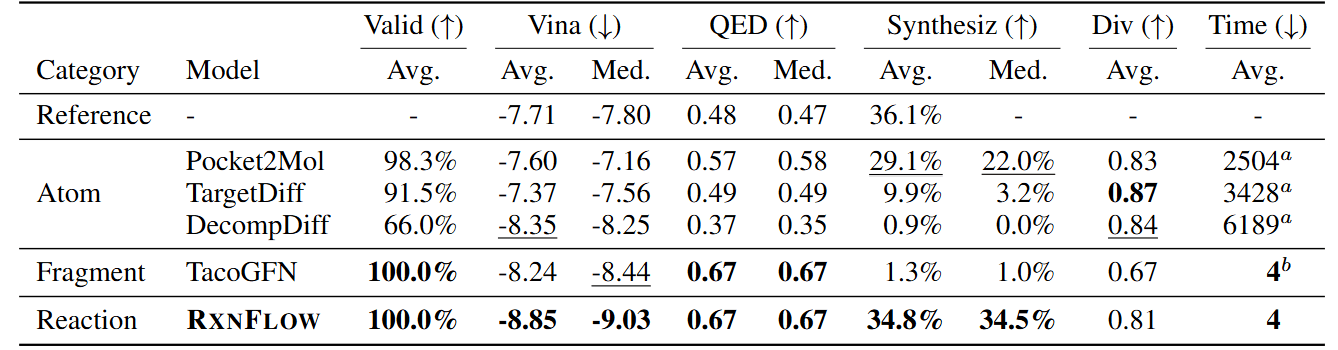
By employing the methods described above, RxnFlow demonstrated superior performance over existing reaction-based molecular generation models across multiple target pockets. In the CrossDocked2020 benchmark, it produced ligands with an average Autodock Vina score of -8.85 kcal/mol and achieved a synthetic feasibility rate of 34.8%. This demonstrates that RxnFlow generates more effective drug candidates more efficiently than previous models. It also overcomes the limitations of earlier molecular generation approaches, which often suffer from low reliability in their generated molecules.
ICLR 2025 Preview: Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows
In structure-based drug design (SBDD), the dynamic properties of proteins are one of the key considerations. These dynamic properties refer to the structural changes a protein undergoes to optimize its interaction with a bound ligand. This phenomenon is known as induced fit. In this context, the protein structure before binding is called the apo form, while the structure after ligand binding is called the holo form.
Most conventional SBDD approaches assume that protein structures are static to improve computational efficiency, which limits the discovery of truly effective drug molecules. Although molecular dynamics (MD) simulations can capture the dynamic changes in protein molecules, they require significant computational time and resources, making them impractical for large-scale drug discovery processes.
To overcome these limitations, the paper proposes an AI model called DynamicFlow. This model collects protein-ligand complex data in both apo and multiple holo states through MD simulations and learns the conformational changes in the protein pockets where ligands bind.
Key Methodologies
-
MD-Based Apo-Holo Data Generation
Limited protein-ligand datasets have been a major bottleneck in training AI models that capture the dynamic properties of proteins. To overcome this limitation, the authors leveraged the MISATO dataset, which includes MD simulation data for approximately 20,000 protein-ligand complexes.
In this dataset, each MD simulation trajectory defines the protein pocket, establishing the holo-structure. Ligand trajectories from the simulations were clustered based on RMSD to identify representative structures among similar holo conformations for use as training data. The apo structures were generated using AlphaFold2 predictions based on the protein sequence, ultimately enabling the authors to construct a dataset comprising 5,692 apo, holo, and ligand interaction structures.
-
Multi-scale Full-Atom Network
The DynamicFlow model converts the apo state of the target protein into the holo state while simultaneously generating ligands docked within the holo-structure. For this process, the input apo protein structure and the ligand, with added noise, are decomposed at the atomic level and embedded. This approach is referred to as the multi-scale full-atom network, which comprises the following two components:
-
Atom-Level SE(3) Equivariant Graph Neural Network (GNN)
First, the apo protein and the ligand with added noise are represented as graphs, with each atom serving as a node. Then, a graph neural network (GNN) is employed to embed each atom in an SE(3)-equivariant manner (i.e., invariant under rotation and translation). The hidden states of the ligand atoms produced by this network are subsequently used to denoise and generate the binding patterns between the ligand atoms and the protein within the holo-structure.
-
Residue-Level Transformer
The hidden states of the protein atoms from the GNN are aggregated at the residue level. These aggregated features, along with the coordinates of the residue frames and torsion angles, are used as input for a residue-level Transformer network. This network updates the 3D transformations and torsion angles of the residue frames to predict the holo-structure.
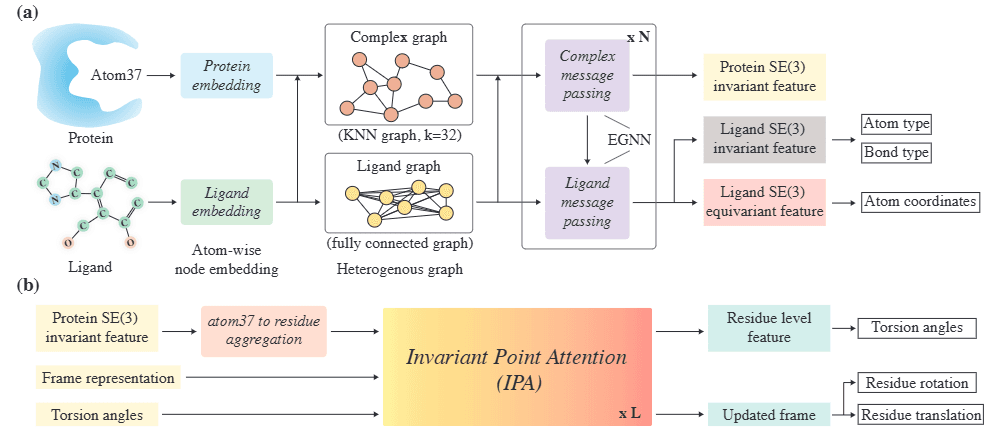
Figure 4. Multi-scale Full-Atom Network Architecture of DynamicFlow [Source] Anonymous authors. Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows. OpenReview. -
Learning Protein–Ligand Dynamic Systems via Flow Matching
The multi-scale full-atom network described above is trained using flow matching. Flow matching is a generative modeling technique that directly learns the transformation path from a simple initial distribution(p_0 ) to the target data distribution(p_1).
In this approach, the model learns to align the vector field (flow) that maps the trajectory from the initial state to the target state. During inference, the model estimates the flow based on the current state and then solves an ordinary differential equation (ODE) to update the state, gradually reaching the target.
In DynamicFlow, the protein’s apo-structure and a ligand with added noise serve as the initial state, while the holo-structure and the docked ligand state act as the target state. These two states undergo interpolation to predefine intermediate states, and the full-atom network is trained to match the flow at each step. During inference, the ODE is solved based on the network’s estimated flow, transforming the apo-structure into the holo-structure while simultaneously generating the docked ligand.
Additionally, the authors introduced DynamicFlow-SDE (Stochastic Differential Equation), where Gaussian noise is introduced into the intermediate states during training. This enhancement results in a more robust model capable of handling high-dimensional noise effectively.
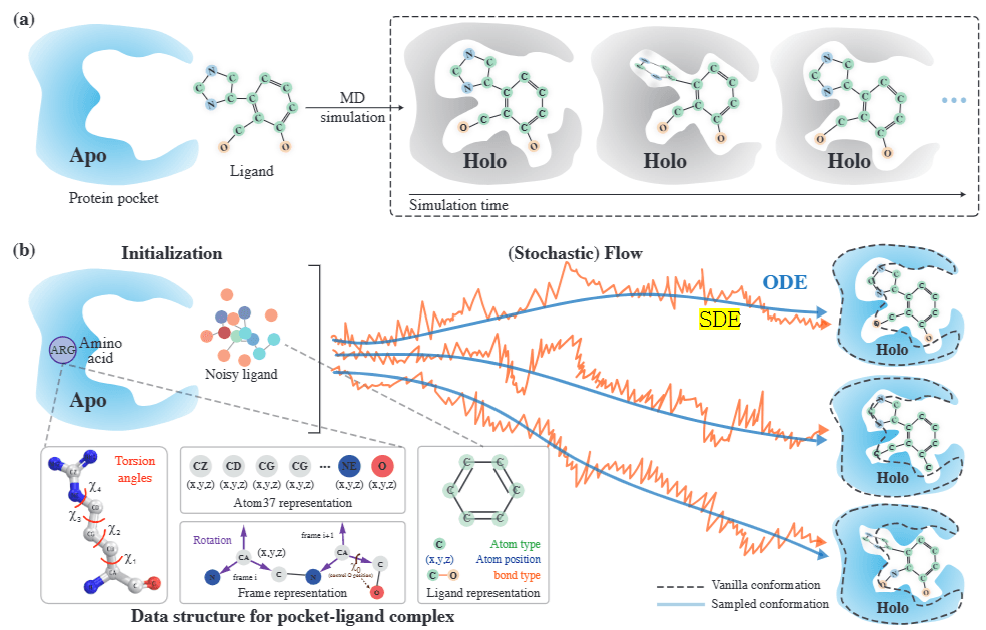
Figure 5. Data Structure of DynamicFlow and Holo Structure & Ligand Generation via Flow Matching [Source] Anonymous authors. Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows. OpenReview.
-
Results
The authors of DynamicFlow evaluated its ligand generation performance using a subset of the MISATO dataset for specific target proteins. In these evaluations, DynamicFlow outperformed conventional SBDD models that rely on static protein structures, producing overall superior ligands.
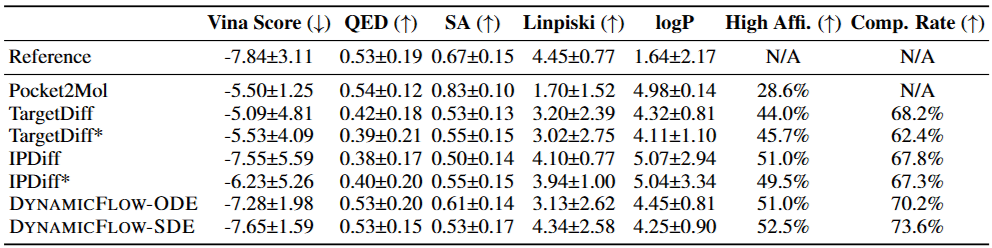
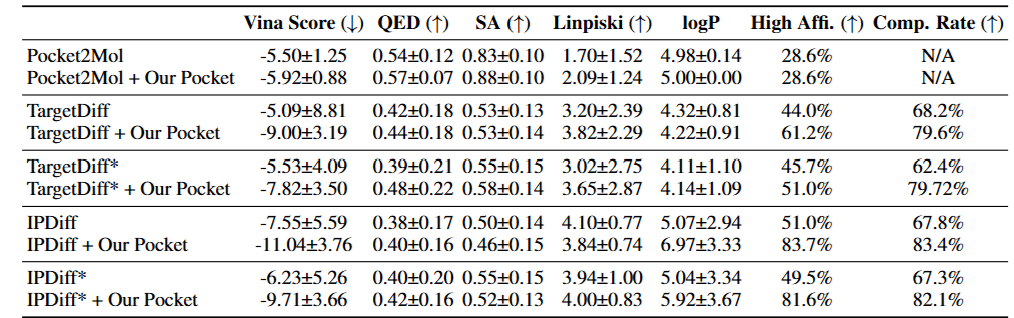
Furthermore, experimental results demonstrated that when DynamicFlow’s holo-structures are integrated into conventional SBDD, superior ligands are generated. In this way, DynamicFlow leverages the dynamic properties of proteins to more effectively identify promising ligands while providing enhanced holo-structure predictions compared to existing models—a breakthrough expected to revolutionize structure-based drug discovery.
ICLR 2025 Preview: NEXT-MOL: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation
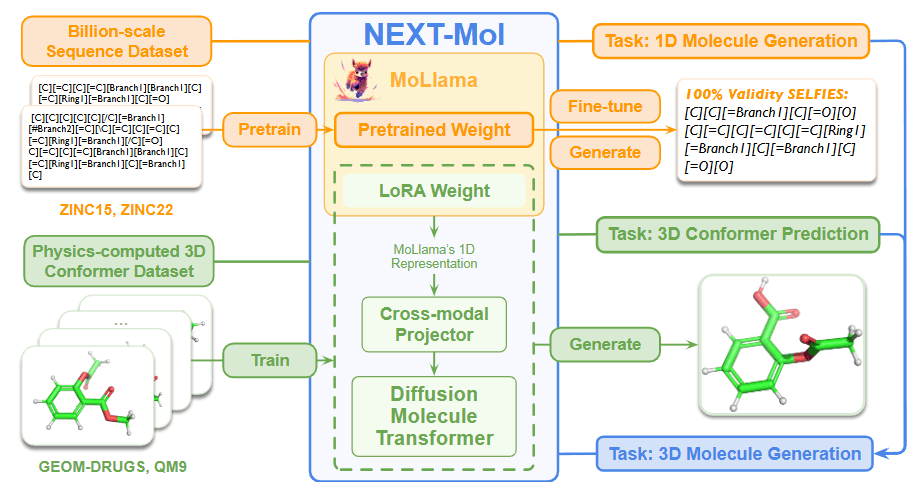
In this preview, we introduce the paper that presents the NEXT-MOL model, which improves 3D molecule generation using a language model that generates 1D molecular representations. The authors point out that previous studies in molecular generation have primarily focused on 3D diffusion models to enhance the performance of molecule structure generation. However, they argue that the advantages of SELFIES-based language models (LMs)—which consistently produce valid molecules and can leverage datasets comprising billions of 1D molecular sequences—have been overlooked.
NEXT-MOL serves as a foundational model for generating 3D molecules. It employs a two-step approach: first, a molecular language model generates 1D molecular sequences (SELFIES), and then a diffusion model predicts the 3D structure (conformer) corresponding to these sequences, ultimately enhancing ligand generation performance.
Key Methodology Description
-
1D SELFIES Generation Language Model (MoLlama)
MoLlama is a language model with 960M parameters that generates 1D molecular sequences through next-token prediction. The authors pre-trained MoLlama using training data comprising 180 million molecules collected from the ZINC-15 database, which were converted into and curated as SELFIES. They also fine-tuned the model via random SELFIES data augmentation, recognizing that even the same molecule can yield different SELFIES when the order of its 2D molecular graph is randomized.
This approach allows MoLlama to be independent of the order of atoms within a molecule, reducing overfitting and enhancing the intrinsic diversity of the molecules encountered during training. As a result of extensive pre-training and fine-tuning, MoLlama captures molecular patterns—such as scaffolds or fragments—that prove useful for subsequent 3D conformer prediction.
-
3D Structure Prediction Model (DMT)
The Diffusion Molecular Transformer (DMT) is used to predict 3D conformers from the 1D SELFIES sequences generated by MoLlama. DMT is built using a Relational Multi-Head Self-Attention (RMHA) network. This architecture simultaneously updates the representations of individual atoms and the relationships between atom pairs, effectively embedding the entire molecule.
DMT employs a diffusion process whereby noise is gradually added to the original data \(x_0\)to transform it into a noisy state \(x_T\), and the reverse process is learned by the neural network. During inference, the network takes a noisy input and progressively removes the noise, ultimately generating data with the desired properties. DMT was pre-trained on datasets such as GEOM-DRUGS and QM9, which contain 3D conformer information. As shown in Figure 6. (a), the network is trained to predict and remove the added noise, thereby restoring the original molecule.
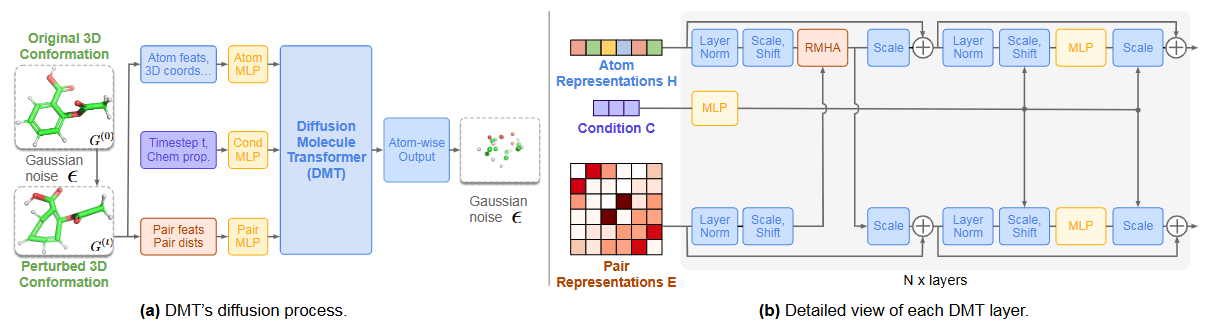
Figure 6. Diffusion Molecular Transformer (DMT) Network Architecture [Source] Anonymous authors. NEXT-MOL: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation. OpenReview. -
Transfer Learning
NEXT-MOL improves overall 3D ligand generation performance through transfer learning, wherein the 1D SELFIES information generated by the pre-trained MoLlama is transferred to the pre-trained DMT.
During this process, the target molecule is represented as a 1D embedding via MoLlama and simultaneously converted into a 2D graph-based input for DMT. A cross-modal projector network is introduced and jointly trained to convert MoLlama’s 1D embedding into a format that DMT can interpret.
The overall transfer learning process is shown in Figure 7 (b). First, as mentioned earlier, DMT is pre-trained using data with 3D conformer information. Then, the pre-trained MoLlama and DMT are linked via the cross-modal projector. With DMT’s parameters fixed, the LoRA network attached to MoLlama and the cross-modal projector undergoes training for a few epochs. The authors refer to this phase as "projector warm up," as it prevents the new cross-modal projector's randomly initialized parameters from disrupting the pre-trained parameters of DMT. Once the projector warmup is complete, NEXT-MOL fine-tunes MoLlama (including its LoRA), the cross-modal projector, and DMT together, thus completing the transfer learning process.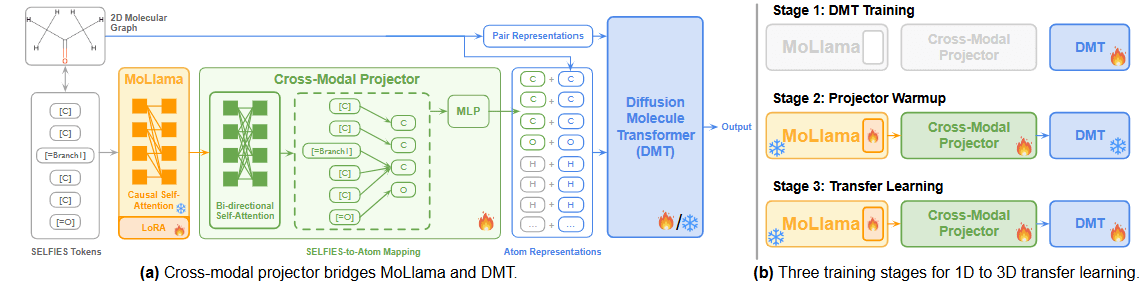
Figure 7. Cross-Modal Projector Architecture and Transfer Learning Process [Source] Anonymous authors. NEXT-MOL: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation. OpenReview.
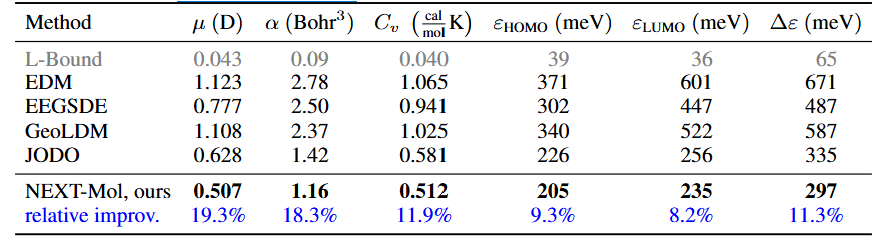
Results
After completing pre-training and transfer learning, NEXT-MOL first generates 1D SELFIES using MoLlama, then predicts conformers with DMT to ultimately generate 3D ligands. Various experiments demonstrated that NEXT-MOL enhances the 3D molecular generation and structure prediction performance of the DMT model. Leveraging a 1D molecular generation language model enables broader utilization of molecular data and demonstrates its potential as a foundational model for novel compound generation.
ICLR 2025: The Future of AI-Created Drug Discovery
Today, we explored some of the most notable AI-driven drug discovery studies presented at ICLR 2025. These studies highlight a key trend in AI-powered drug design: leveraging generative modeling techniques such as Diffusion and Flow Matching to create structure-based molecular generation methods that effectively bind to target proteins.
Various approaches have been applied to enhance the synthetic feasibility of generated molecules. RxnFlow employs building blocks and reaction templates to learn sequential molecular synthesis processes, while DynamicFlow incorporates MD simulations to account for the induced fit of protein-ligand binding. Additionally, NEXT-MOL utilizes pre-trained 1D molecular sequence language models to compensate for limited 3D conformer data. These advancements have significantly improved the quality of AI-generated molecules.
Through this year’s ICLR, it has become evident that AI is evolving beyond merely "predicting" the properties of drug molecules or protein structures—entering the realm of "creating" entirely new drugs. The era of AI-driven drug discovery is no longer a distant future; it is rapidly becoming an inevitable reality. How will AI continue to revolutionize drug discovery? And how will this transformation impact global health and human longevity? The future holds exciting possibilities.
Amidst this shift, Hyper Lab is playing a crucial role in making AI-driven drug discovery more efficient. Our AI-powered platform enables rapid identification of promising drug candidates while being designed for ease of use by researchers. A free trial is also available—so experience the potential of AI in drug discovery with Hyper Lab today!
Start your Free-trial: https://buly.kr/AapClS1
Schedule an Meeting: https://abit.ly/6tr1uz