Hyper-Binding Co-Folding: Innovations in Drug Development After AlphaFold 3


Hello, this is Jaechang Lim, CTO of HITS.
As AI technology continues to advance at a rapid pace, its applications across various research domains are becoming increasingly promising. In particular, the introduction of AlphaFold has revolutionized the fields of life sciences and medical research. Now, its successor, AlphaFold3, is garnering even greater attention.
However, drug discovery is a complex and highly precise process that demands more than just static structure prediction. To truly empower researchers, we need advanced technologies capable of accurately predicting molecular interactions. This is why HITS has developed Hyper Binding Co-folding—an AI-powered technology designed to deliver performance and usability that researchers can tangibly experience in real-world applications.
In this article, I will explain how AlphaFold3 represents a significant technical advancement and how our Hyper Binding Co-folding technology builds upon this foundation to enhance the accuracy and efficiency of drug discovery.
AlphaFold3 & Molecular Docking: A New Frontier for Drug Discovery
Published in Nature in 2024, AlphaFold3 has achieved remarkable success in predicting interactions not only between proteins but also with DNA, RNA, ligands, and more. While AlphaFold2 primarily focused on protein structure prediction, AlphaFold3 expands its capabilities to include binding pose predictions, nucleic acid interactions, covalent modifications, and beyond—surpassing traditional tools like AutoDock Vina and RoseTTAFold in many key benchmarks.
Although computer-aided drug design (CADD) tools are increasingly adopted by experimental researchers, current docking methods still face significant limitations. AlphaFold3’s advancements provide innovative solutions to many of these longstanding challenges.
Three Major Limitations of Traditional Docking Methods
- Neglect of Protein Flexibility:
- Proteins are not rigid; they’re dynamic molecules. When ligands bind, proteins can undergo structural changes (known as induced fit), which significantly affect binding affinity.
- Most conventional docking tools treat proteins as rigid or struggle to model flexibility accurately due to the complexity of simulating thousands of atoms.
- While molecular dynamics (MD) simulations can account for flexibility, they are computationally expensive and require expert configuration—making them inaccessible to many experimental researchers.
- Difficulties Predicting Binding Sites:
- Drugs can theoretically bind anywhere on a protein surface, but searching all possibilities is computationally infeasible. Most tools require users to specify a binding site.
- When binding sites are unknown, blind docking leads to lower accuracy and significantly longer computation times.
- Dependency on Input Structure Quality:
- Docking accuracy heavily depends on the quality of the protein’s 3D structure:
- Only sequence available: Traditional docking isn’t applicable unless a homology model is built, which may lack precision.
- Apo structure only: Predictions are less reliable, and most new targets fall into this category.
- Holo structure available: This is ideal, but rarely available for novel targets.
- Docking accuracy heavily depends on the quality of the protein’s 3D structure:
This structural uncertainty presents a major challenge, especially when developing "first-in-class" drugs for new targets.
Hyper Binding Co-folding: A New Docking Paradigm
To overcome these limitations, HITS extended the AlphaFold3 approach to develop Hyper Binding Co-folding, a novel docking technology. It predicts complex structures directly from the sequence information of proteins, RNA/DNA, and ligands—enabling a co-folded view of the entire binding interaction.
Here are four core advantages of Hyper Binding Co-folding:
- Precise Induced Fit Modeling: By generating protein structures optimized for each ligand from sequence data, it captures fine conformational changes critical for accurate predictions.
- No Need to Predefine Binding Sites: Unlike traditional tools, Co-folding predicts binding without requiring prior knowledge of the site. Supplying site information can further enhance accuracy, but it’s optional.
- Simplified Input Preparation: No need to hunt for the “best” protein structure. Just select the protein sequence—Hyper Binding Co-folding handles the rest.
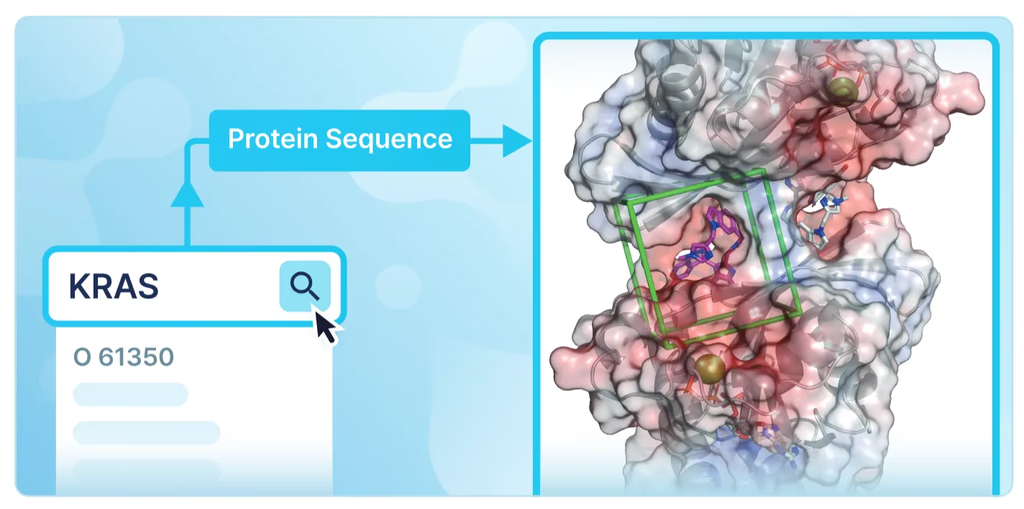
- Exceptional Ease of Use: Experimental researchers often find structure prep to be a bottleneck. With intelligent search for protein names (e.g., "KRAS") and automatic retrieval of metadata (UniProt ID, species, gene info), setting up your input has never been easier.
Typical workflow:
- Enter protein name (Auto-search by HITS database)
- Review/edit sequence, cofactors, metal ions, PTMs
- Input ligand 2D structure (Conformer generated automatically)
- Predict binding structure and binding affinity
Performance & Applications of Hyper Binding Co-folding
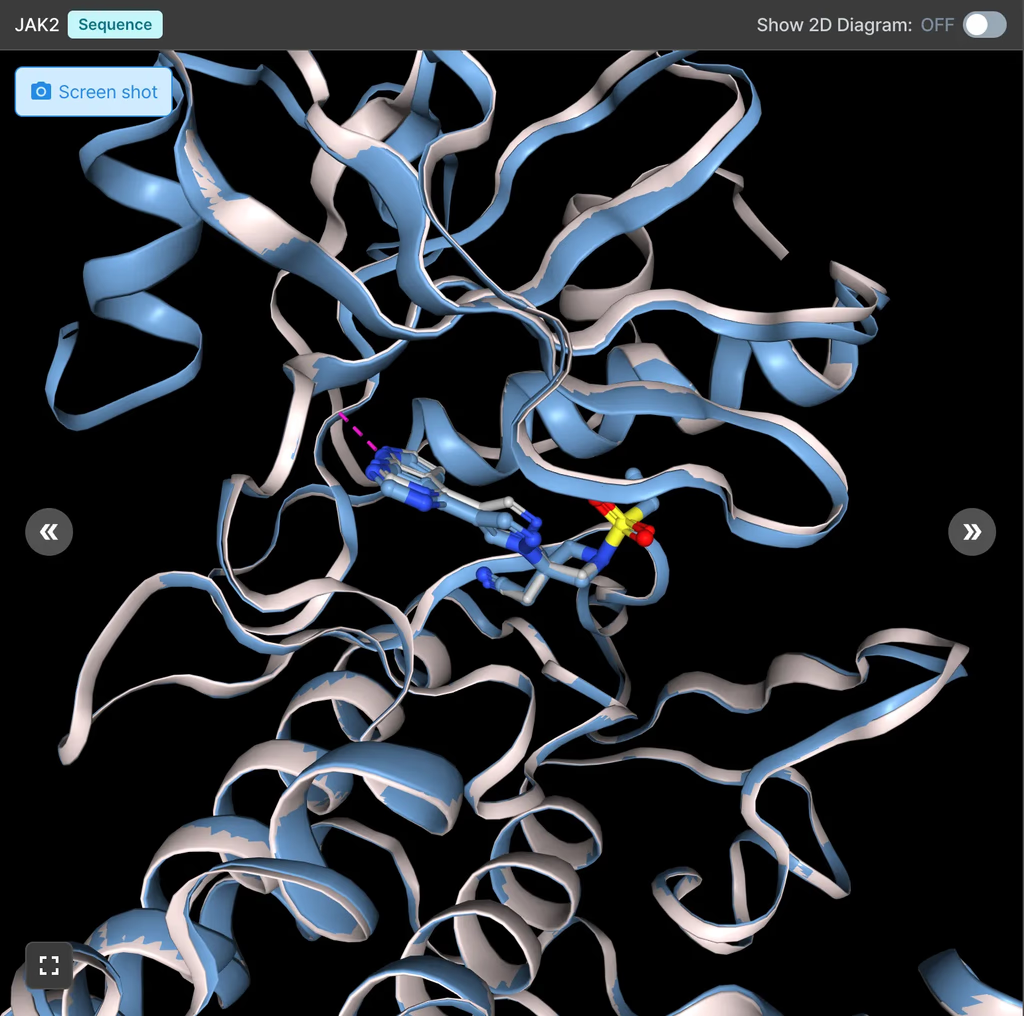
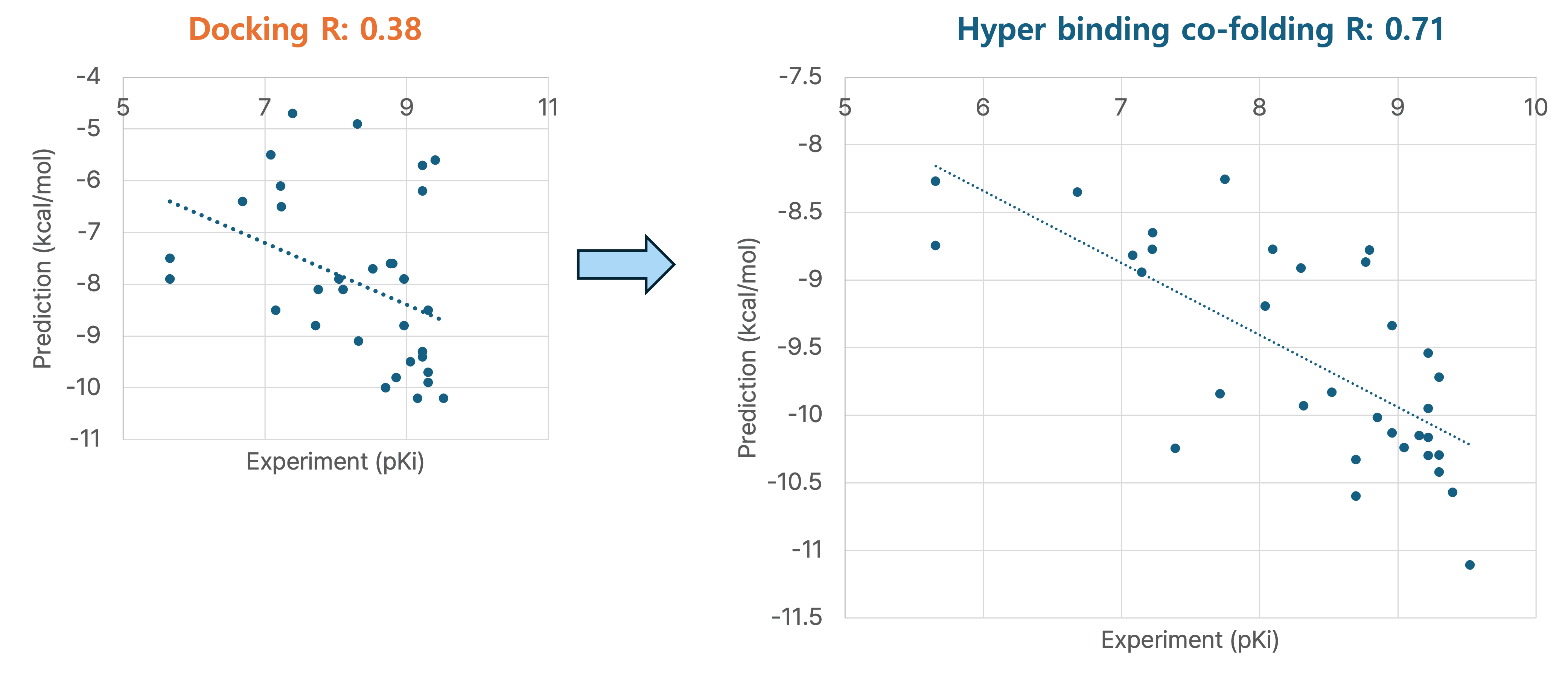
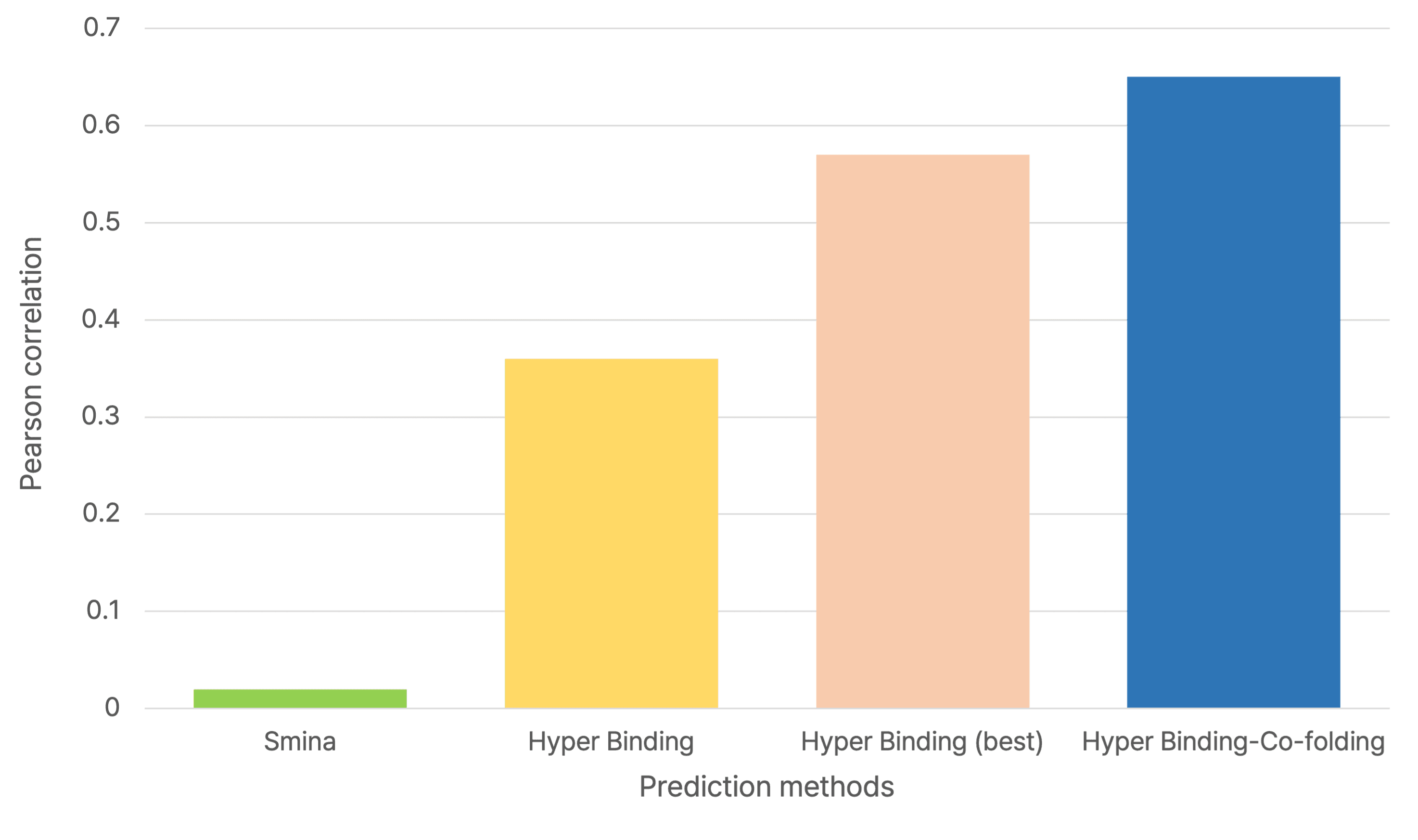
1. JAK2 Inhibitor Binding Prediction
Using Hyper Binding Co-folding, the Pearson correlation (R) with experimental pKi values improved from 0.38 to 0.71, showing a significant gain in predictive power.
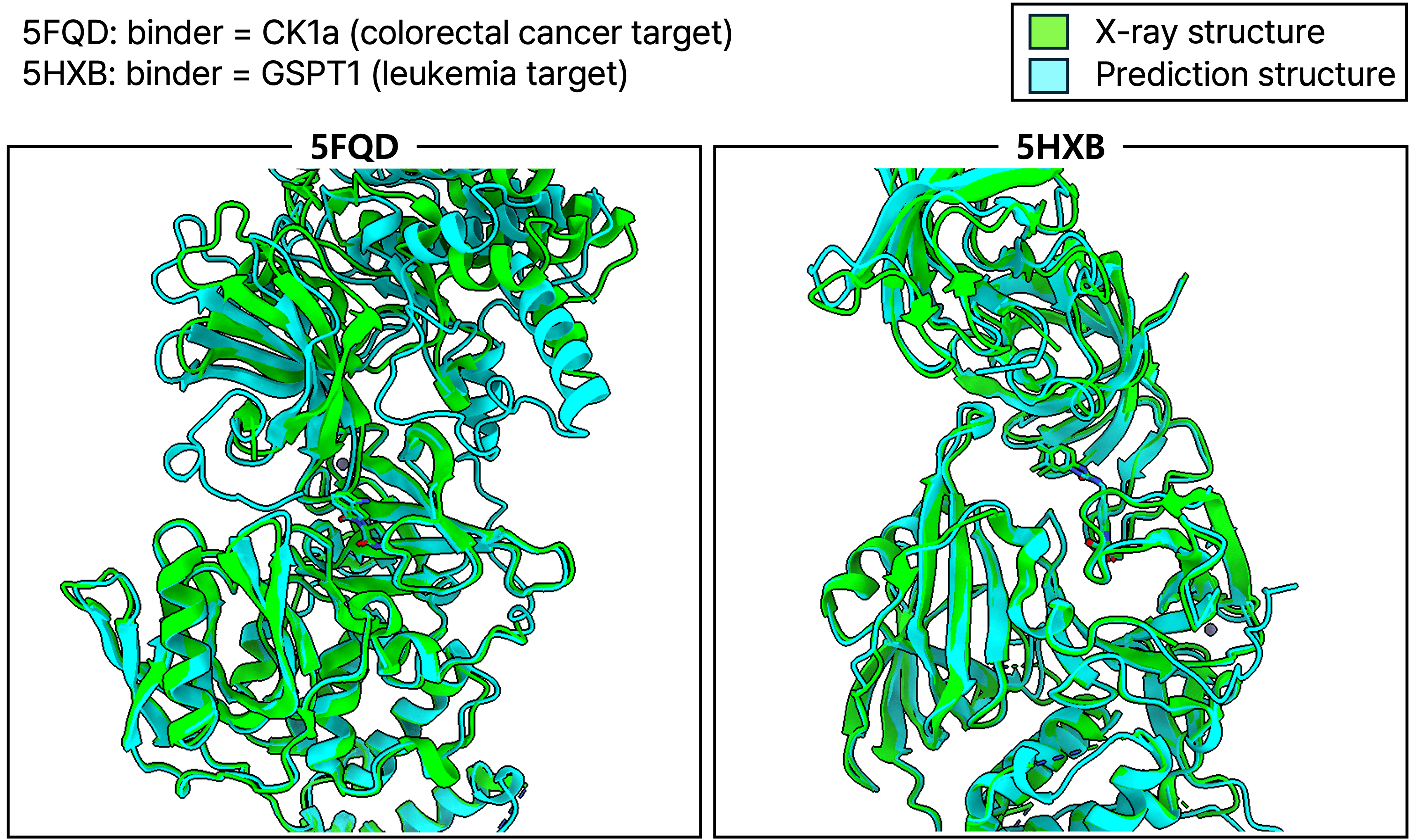
2. Molecular Glue Prediction
This technology also enables accurate prediction of ternary complexes, such as those formed by molecular glues—small molecules that induce protein-protein interactions to trigger target degradation. In cases like CK1a (PDB: 5FQD) and GSPT1 (PDB: 5HXB), predicted structures closely matched experimentally determined complexes—even capturing structural shifts caused by the presence of the molecular glue.
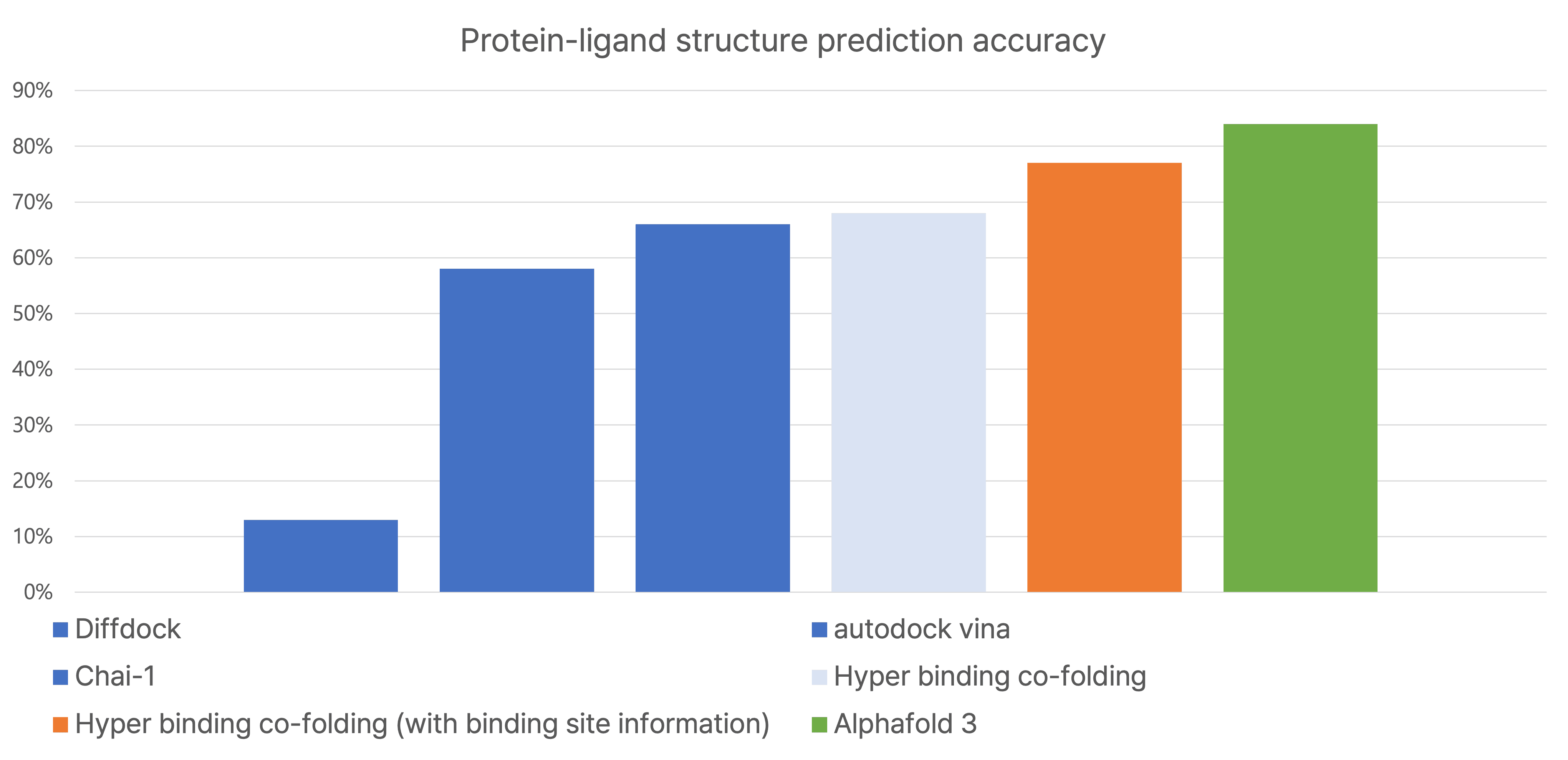
3. Accuracy Comparison
Hyper Binding Co-folding outperformed traditional tools and even AlphaFold3 on PoseBusters v2, especially when binding site information was used. It also showed higher predictive accuracy for ligand-protein activity using only sequence data, surpassing models using ideal Holo structures.
4. User Experience Comparison
| 기능 | Hyper Binding Co-folding | AlphaFold3 | Chai Discovery |
|---|---|---|---|
| Induced fit modeling | O | O | O |
| RNA prediction | O | O | O |
| DNA prediction | O | O | O |
| Protein keyword search | O | X | X |
| Binding site guidance | O | X | X |
| Preprocessed site suggestions | O | X | X |
| Binding affinity prediction | O | X | X |
Try Hyper Binding Co-folding Free for One Week
Hyper Binding Co-folding was designed to empower researchers of all backgrounds, even those without deep expertise in structural biology, cheminformatics, or molecular design. It’s particularly well-suited for:
- Experimental researchers without 3D structure data
- Early-stage teams exploring novel targets or first-in-class drugs
- Labs focused on molecular glues, PROTACs, or complex drug modalities
- Anyone who’s struggled with the accuracy or usability of traditional docking tools
By combining powerful AI with a user-friendly interface, Hyper Binding Co-folding enables accurate, high-throughput predictions using only a protein sequence and ligand structure—no complex prep or assumptions required.
The technology is available now on the HyperLab platform, with a free one-week trial for all users.
Discover a simpler, smarter way to approach structure-based drug design. Experience the future of docking today—start your free trial on HyperLab.
Start your Free-trial: https://buly.kr/612Ar3U
Schedule an Meeting: https://abit.ly/6tr1uz