Protein Structure Based Drug Design - Docking Environment Setup


In molecular modeling specifically, molecular docking aims to predict stable binding structures by analyzing the interactions between two molecules. However, in drug discovery, it is primarily used to indicate the binding structures between compounds and target proteins. In this context, compounds are called ligands, and target proteins are called receptors.
Successful docking requires consideration of protein structure selection, binding site definition, and environment settings. In this article, we will explore the environmental settings of docking programs.
Components of Docking Programs
To understand the environment settings of docking programs, let me briefly introduce the components of docking.
Components of Docking Programs
To understand the environmental settings of docking, it’s helpful to first outline its core components.
1. Docking Algorithm
- Generates various ligand-receptor binding conformations, which are evaluated using a scoring function.
- Rigid Docking: Assumes fixed conformations for the ligand and receptor, rotating and translating the ligand for docking.
- Flexible Docking: Accounts for ligand conformational changes and sometimes allows flexibility in the receptor’s side chains. Ligand flexibility is often handled by torsional angle changes, with some programs even considering ring structure variations.
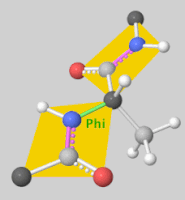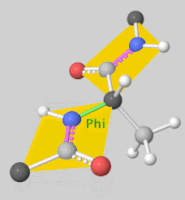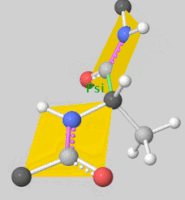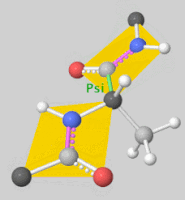
2. Scoring Function
- A mathematical model is used to calculate binding affinity.
- Physical-based scoring functions: Use force fields to compute interactions, considering hydrogen bonds, electrostatics, van der Waals forces, and entropy.
- Empirical scoring functions: Trained on known protein-ligand structures and binding affinity data, offering fast calculations but limited accuracy outside the training data.
- Consensus scoring functions: Combine scores from different scoring functions, though low discriminatory power can arise if correlated functions are used.
3. Optimization Algorithm
- Finds variable values that minimize the loss function (typically the scoring function). Variables include torsional angles, center, and rotation angles defining ligand conformation and position.
- Optimization techniques such as genetic algorithms, simulated annealing, and random walks are commonly employed to achieve stable ligand-receptor binding structures.
Configuring Docking Environments
Docking environment settings are closely related to the components mentioned above. Appropriate configurations are crucial for achieving a balance between accuracy and computational efficiency.
1. Thoroughness (Exhaustiveness)
- Defines the extent of ligand-receptor binding structure exploration during docking. Higher thoroughness improves accuracy and reproducibility but subsequently increases computation time.
- The number of rotatable single bonds in a molecule determines its flexibility. For example, a molecule with three rotatable single bonds can have (360°/30°)3=1728 conformations if rotated in 30° increments. Low thoroughness may sample only a subset of these, risking failure to find the optimal conformation. (360°/30°)3=1728(360°/30°)^3 = 1728
- Conversely, excessive thoroughness can waste computational resources, especially during large-scale docking. Setting appropriate values through testing is essential.
2. Number of Binding Poses
- Docking programs usually produce approximately ten different binding poses, each with an associated score. While this number is often adequate, highly flexible ligands or docking scenarios involving receptor flexibility may necessitate the review of additional poses.
- One effective way to determine the suitability of binding poses is to evaluate critical interactions. For instance, kinase inhibitors typically require the formation of hinge-binding interactions, which can serve as an important criterion. Tools such as Hyper Lab’s 3D viewer provide a convenient platform for detailed analysis of binding pose quality.
3. Scoring Function & Optimization Algorithm
- Choosing an appropriate scoring function and optimization algorithm is essential for improving prediction accuracy. Physics-based scoring functions are particularly effective for predicting molecular structures, while recent machine learning approaches, trained on X-ray crystallography data, demonstrate significant advantages in activity prediction.
- Hyper Lab’s Hyper Binding integrates physics-based methods and machine learning techniques to deliver exceptional performance in predicting activity and structural conformation.
4. Flexible Residue Settings
- When receptor flexibility plays a crucial role, docking simulations should account for it. Ligand binding that causes substantial structural rearrangements in the receptor may require the use of several distinct receptor structures. For residues whose conformations vary depending on the ligand, it is possible to include flexible adjustments during the docking process.
- Incorporating flexible residues increases the search space, which can result in longer computation times and reduced accuracy. Therefore, thorough testing and careful evaluation of strategies are essential.
By now, you should have a general high-level understanding of configuring the docking environment for protein-drug binding structure prediction. It is essential to carefully analyze and apply docking results with a critical perspective. In the next article, we will discuss the inherent limitations of docking and strategies for effectively utilizing its outcomes.