How to discover and define binding sites in structure-based drug discovery


Structure-Based Drug Design (SBDD) Overview
Structure-Based Drug Design Definition
Structure-Based Drug Design (SBDD) is a method that uses the three-dimensional structure of a specific biological target (usually a protein) in the drug discovery and development process. SBDD plays an important role in developing new drugs more quickly and efficiently. It is widely used to develop treatments for a variety of diseases, including cancer, infectious diseases, and neurological disorders.
Basic process of structure-based drug discovery
Structure-based drug discovery requires the following preparations.
- Selecting the three-dimensional structure of the target protein
- Preparation of compound library
The three-dimensional structure of a target protein can usually be found in the RCSB. In the process of selecting the protein structure, the most important thing to pay attention to is the binding site (or binding pocket). Structure-based drug discovery uses docking to predict the three-dimensional binding structure of a compound with a target protein, and the location on the protein where the drug binds is called the binding site. The definition of binding site is a key process that determines the outcome of structure-based drug discovery research, so we will cover it in detail.
Defining a binding site in structure-based drug discovery
When defining a binding site, you must first clarify the mechanism of action (MOA) of the drug you want to develop. Depending on the mechanism of action of the drug, the location, size, structure, and composition of the binding site will be different even for the same target protein. Let's talk about how to define the binding site in different situations.
Choosing binding site location and size
A protein can have more than one binding site. For example, in the case of kinases, the ATP binding site is the most widely used for the development of competitive inhibitors, but it is known that there are various other allosteric sites that can be targets for drug discovery. The figure below shows the known allosteric sites of kinases.
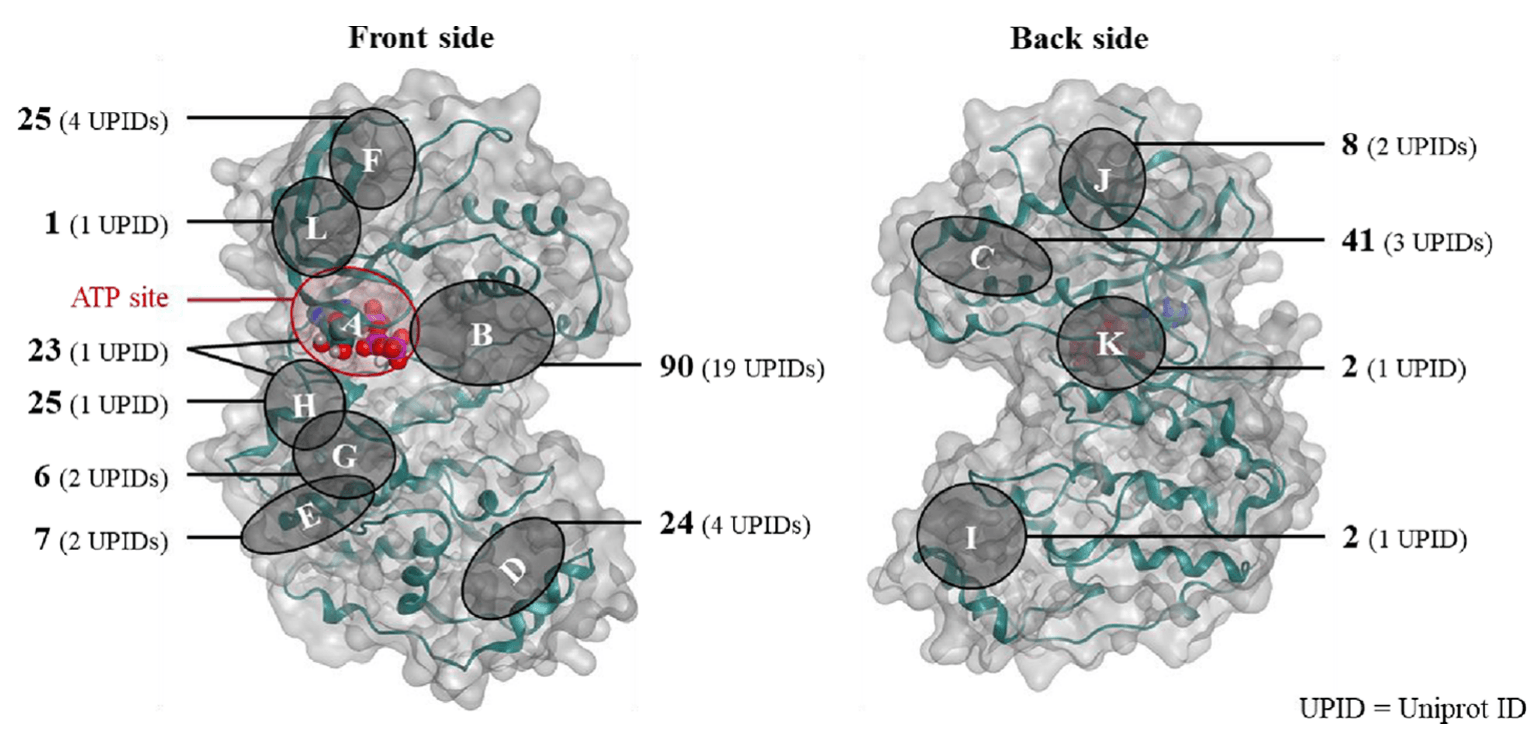
Hyper Lab, an AI drug discovery platform, automatically selects a ligand if it is included in the protein structure (PDB file) you select, and defines a binding site around the ligand. However, if the ligand binds to a different binding site than the one you have selected, you will not get the desired result. Therefore, even if you have selected a target protein, you need to check which binding site is suitable for your planned MOA and, if possible, find a structure where the drug binds to that binding site.
Selecting a binding site structure
In their experimentally determined structures, such as X-ray or Cryo-EM, proteins look like fixed, three-dimensional structures. However, proteins are fluid structures that move around in vivo, undergoing large and small changes due to the binding of cofactors, substrates, inhibitors, etc. The figure below shows the behavior of a protein predicted by Molecular Dynamics (MD). From an SBDD perspective, this leads to the problem of protein structure selection.
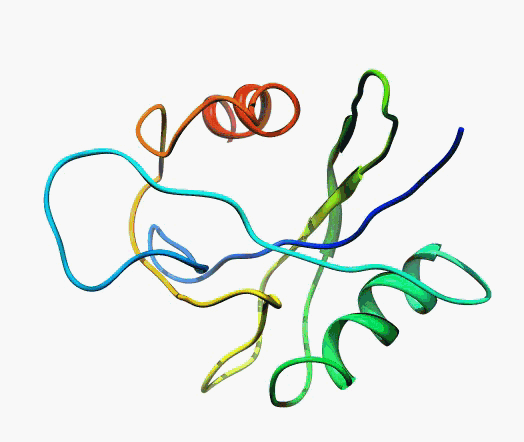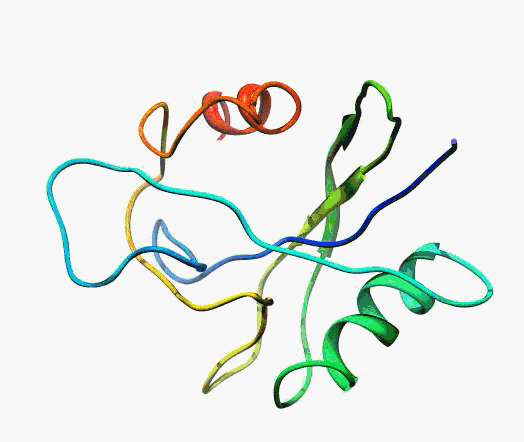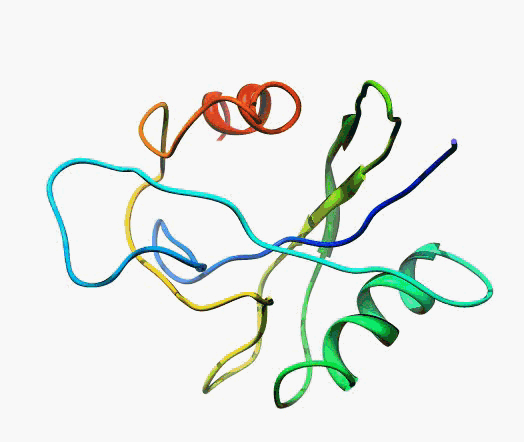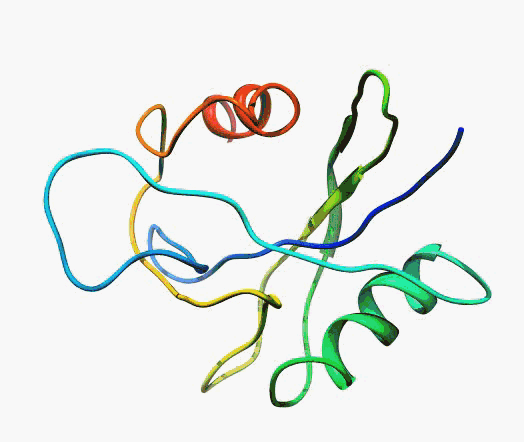
The function and activity of a protein is determined by its structure. In simple terms, the structure of a protein determines its function as a receptor, enzyme, or transporter, and the unique structure of each of these proteins is determined by its amino acid sequence. In addition, proteins undergo structural changes due to binding of substrates or drugs or modifications such as phosphorylation and methylation, which leads to changes in protein activity.
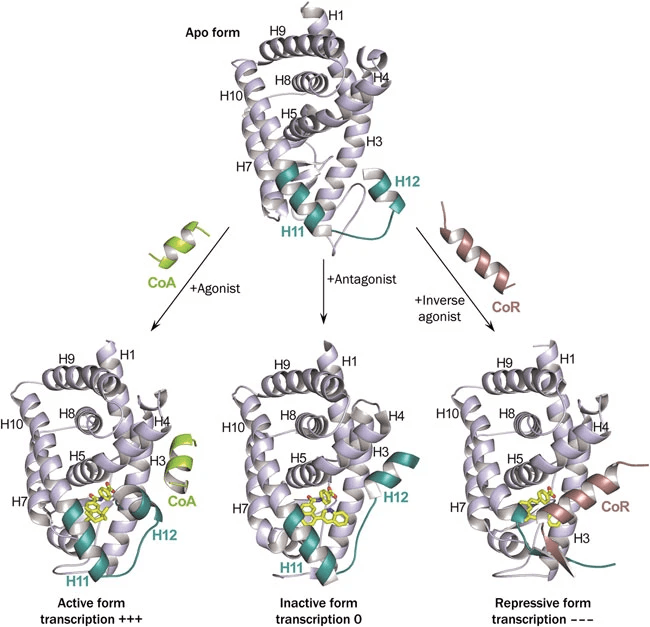
The figure below shows the structure (specifically H12) and activity changes of a nuclear receptor when an agonist, an antagonist, and an inverse antagonist bind to the same binding site of a nuclear receptor (NR), respectively.
(Left) When agonist is bound, H12 is in a stable active position, allowing it to interact with transcriptional coactivators.
(Center) In the case of Antagonist, it occupies the space where H12 in the active postion would be located, preventing H12 from achieving this active position.
(Right) Finally, inverse agonists stabilize the interaction with the corepressor (CoR), which inhibits the activity of NR.
Intuitively, if you want to develop an agonsit, you should use a structure in the active conformation. If you use a structure that has an antagonist bound to it, you will likely end up with a structure that is inadvertently less active.
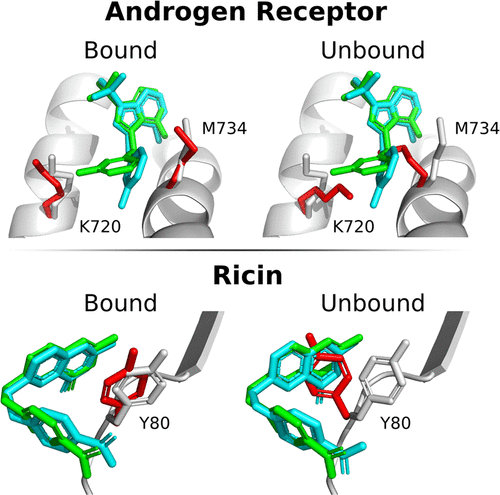
As shown above, the structure of a protein can be different when a drug binds to it than when it doesn't. For the same type of drug, say an inhibitor, will the structure of the target protein it binds to be the same?
This depends on the structural features (flexibility) of the protein, but in general, if the structure of an inhibitor is different, there may be structural changes in the side chains at a small scale and in the backbone at a larger scale. In the figure below
(Top) androgen receptor and ricin when ligands of different structures bind to them and
(Bottom) the structural differences in the side chains of androgen receptor and ricin when ligands of different structures are bound.
While most side chains have similar conformations, some, especially the flexible Met, can vary greatly in conformation, which can be a very important variable in predicting the binding structure of proteins and drugs. Therefore, when conducting SBDD studies, it is important to look at the structures of as many target proteins as possible to understand their conformational fluidity.
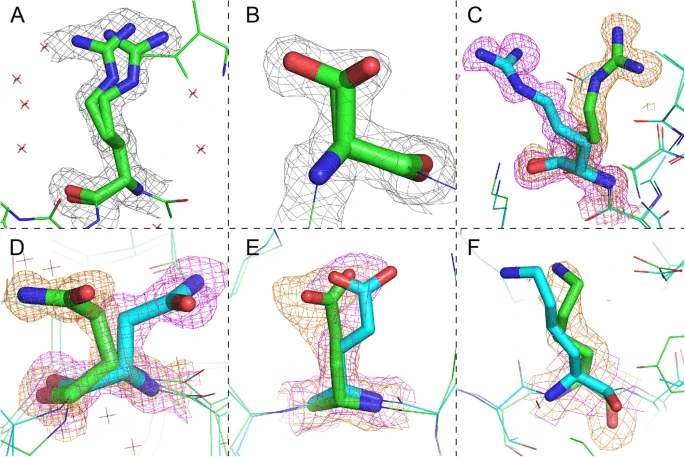
Note that Alternative Conformations are present in the PDB file. As mentioned earlier, proteins are dynamic structures, so for side chains with large motions, there may be more than one structure in the PDB. Due to the large movement, selecting the wrong conformation can act as an obstacle to obtaining the correct protein-drug binding structure. Hyperlab, an AI drug discovery platform, automatically selects the conformation with the highest probability of existence.
Binding site configuration selection
For typical competitive inhibitors, the binding site can be defined as the amino acids of the target protein that interact with the drug. However, if the drug interacts with components other than amino acids, a strategy is needed to represent this appropriately.
In the case of interacting with a cofactor, the presence or absence of the cofactor will affect the activity of the drug, and this needs to be reflected appropriately in the docking process. For example, consider the figures below.
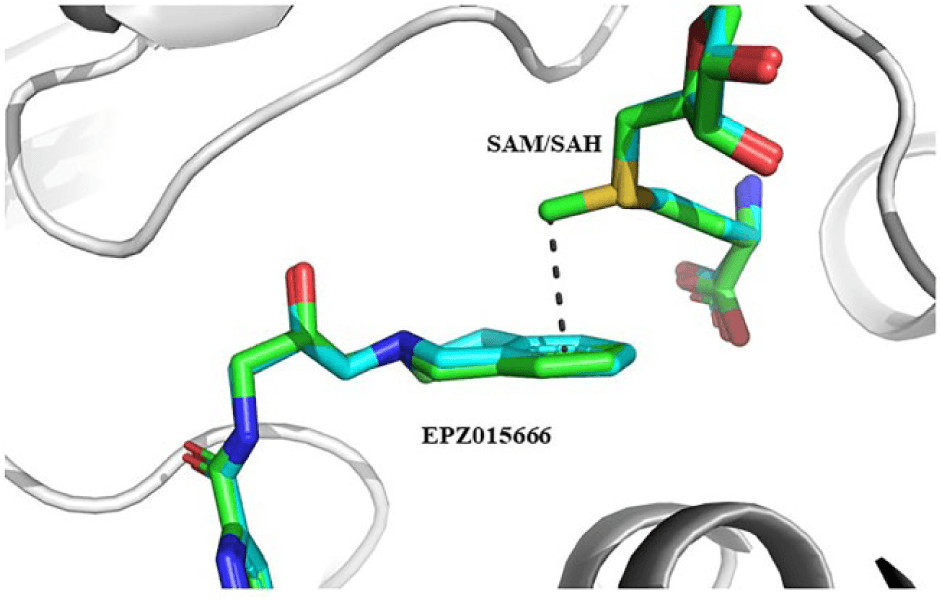
(First figure) EPZ015666, an inhibitor, has a pi-cation interaction with SAM, a coenzyme.
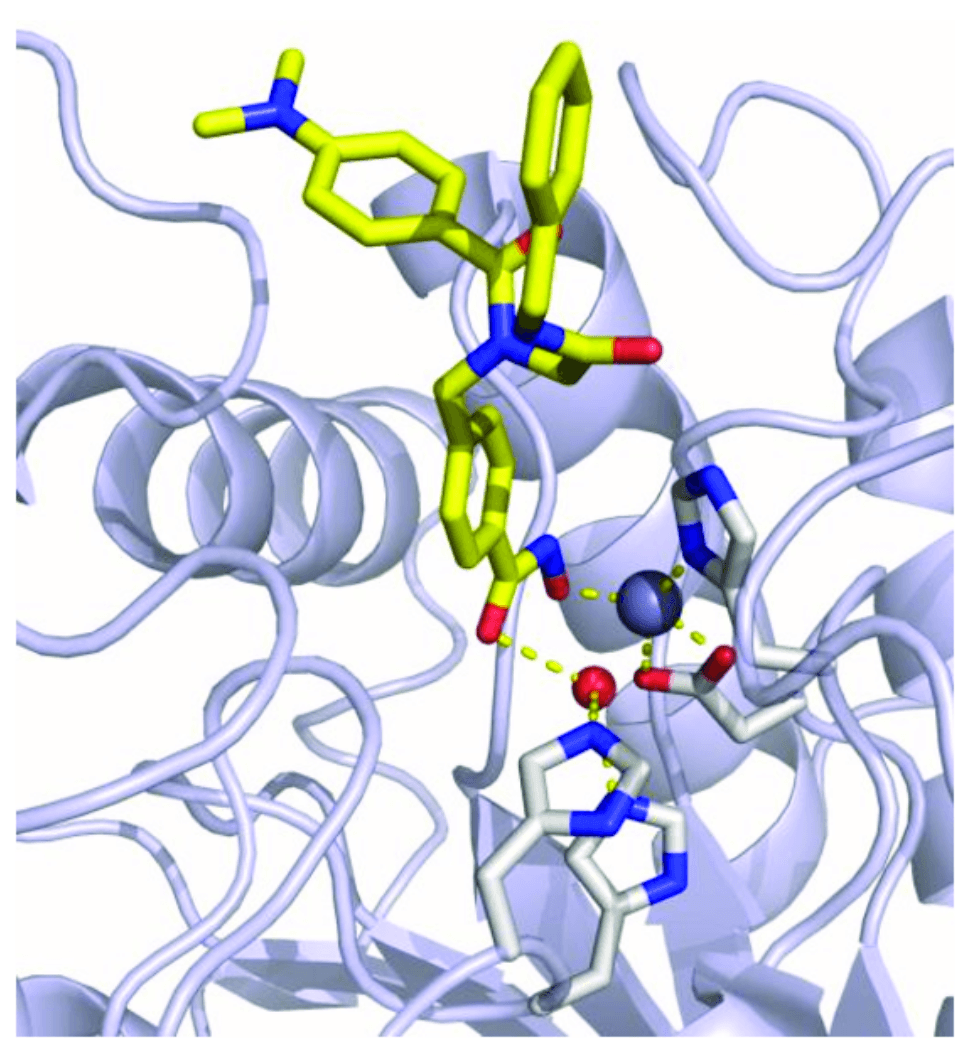
(Second figure) We can see that the inhibitor DDK-137 coordinates with the ZN ion.
In this case, these cofactors should be selected as part of the binding site to get the right result. In the AI drug discovery platform Hyperlab, this can be solved by selecting the cofactors as part of the receptor during the binding site definition process.
Some inhibitors have special interactions in addition to non-bonding interactions such as hydrogen bonding or van der Waals. Examples include covalent inhibitors that have covalent bonds with residues such as Ser or Cys in the binding site, or inhibitors that have coordination with metals such as Zn or Fe. For these special interactions, a docking method must be applied to properly account for them.
Wrapping up
In today's article, we introduced the definition and basic process of structure-based drug design (SBDD). Structure-based drug design (SBDD) is a process of understanding the three-dimensional structure of proteins and designing effective drug candidates based on it. This approach enables more accurate and effective drug development by precisely analyzing the structure and function of target proteins involved in specific diseases. We hope this article has helped you better understand binding sites, which play an important role in SBDD, through various examples of situations.