Hyper Screening X : 세계 최대 분자 라이브러리로 신약 개발의 미래를 열다


Hyper Screening X: 딥러닝을 이용한 11조 개 초거대 라이브러리 가상 탐색
가상 탐색은 신약개발 초기 단계에서 유효 물질을 찾는 방법으로 널리 활용되고 있습니다. 기술의 발전으로 가상 탐색이 가능한 라이브러리의 크기는 점차 증가하여 수백~수천만에 이르고 있습니다. 라이브러리의 크기가 증가하면 증가할수록 신규 유효물질을 발굴할 확률 또한 증가합니다.
최근에는 초거대 화합물 라이브러리의 등장으로 라이브러리 크기가 기하급수적으로 증가하여 조 단위에 이르고 있습니다. 오늘은 수조 개 분자로 이루어진 초거대 화합물 라이브러리를 효율적으로 탐색하는 방법에 대해서 알아보도록 하겠습니다.
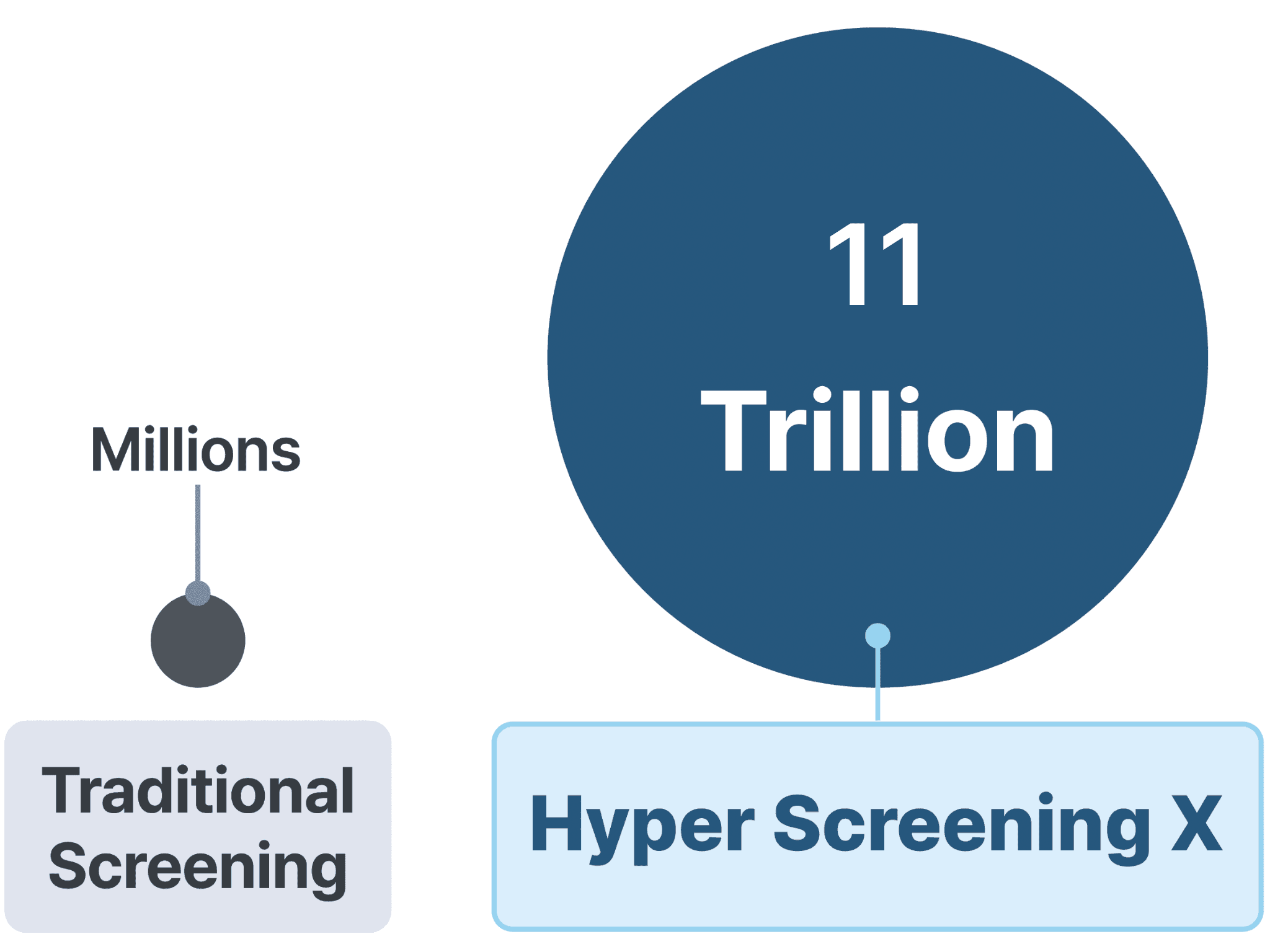
일반 가상탐색 라이브러리 크기 비교 모식도
딥러닝이 필요한 이유
Docking을 이용한 단백질 구조 기반 가상탐색이, 1개의 분자를 계산하는데 10초가 소요된다면 100만 개의 분자를 계산하는 데는 1000만 초, 약 115일의 시간이 소요됩니다. 3달이 넘는 긴 시간이기에 이를 줄이기 위해 여러 컴퓨터를 이용하여 병렬계산을 수행하곤 합니다. (병렬계산 = 동시에 여러 계산을 수행하는 것)
컴퓨터 100대를 이용한다면 이 같은 소요 시간을 1/100로 단축할 수 있습니다. 이상적으로 1.15일만에 계산을 완료할 수 있습니다. 컴퓨터 100대만 있다면 1.15일은 충분히 기다릴 수 있는 시간입니다.
분자가 1,000만개라면 11.5일이 걸릴 것이고, 이 역시도 기다릴 수 있을 만한 시간입니다. 이렇듯 기존 방법으로도 대략 수천만 개 정도의 분자는 계산할 수 있습니다.
하지만 이를 넘어가면 감당하기 어려운 것도 현실입니다.
만약 100억 개의 분자를 가상 탐색한다면? 컴퓨터 1,000대를 이용하더라도 3년이라는 긴 시간이 소요됩니다.
11조 개의 분자를 가상 탐색한다면? 3,000년이 소요됩니다. 가상 탐색을 위해 이 정도의 시간을 기다릴 수는 없겠죠.
이러한 산술적 접근 방식을 통해, 우리는 11조 개나 되는 가상 라이브러리를 탐색하기 위해서, 보다 영리하고 스마트한 방법을 찾아야함을 알 수 있습니다. 다행히도 이를 해결하는 방법이 있습니다. 바로 RxnFlow입니다.
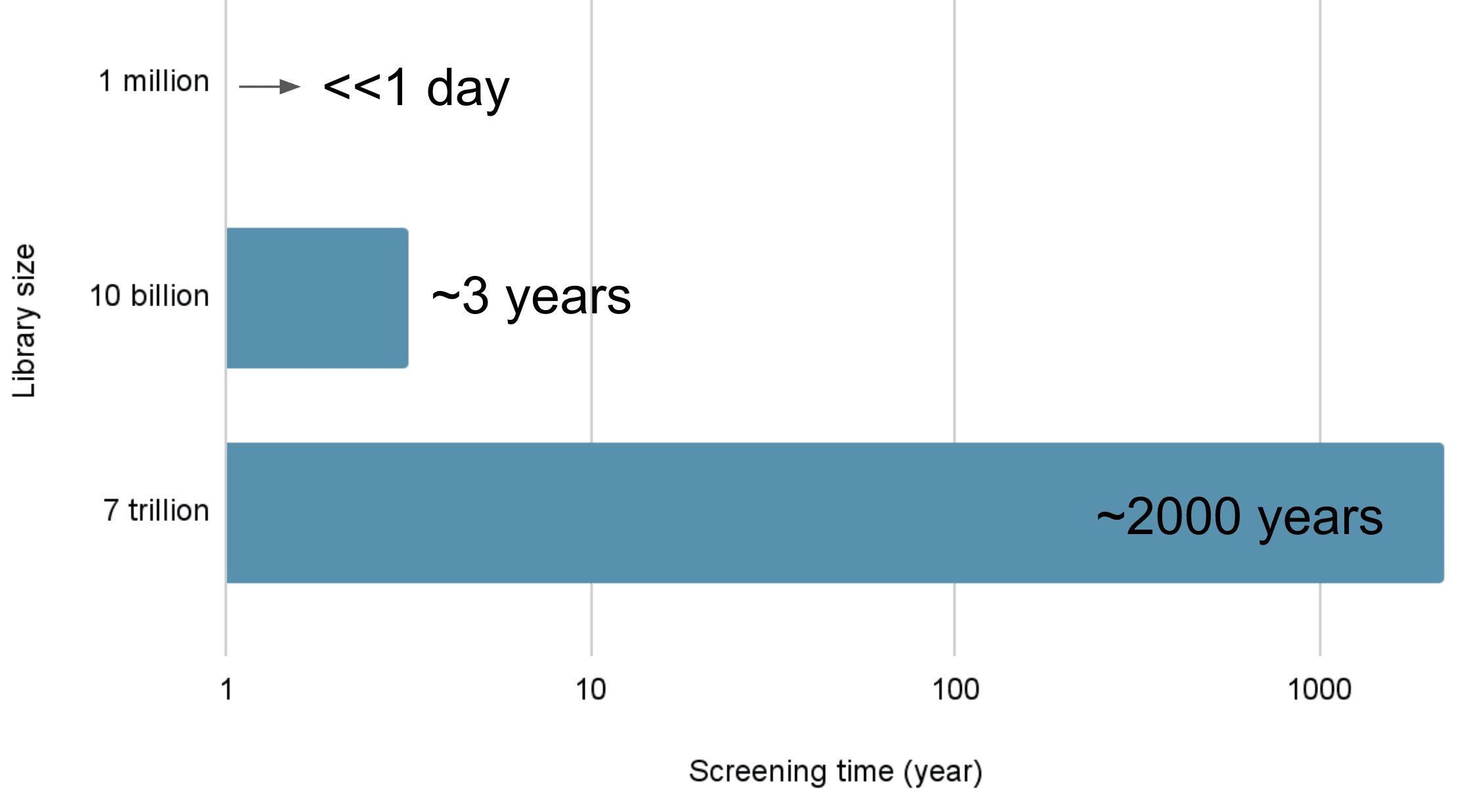
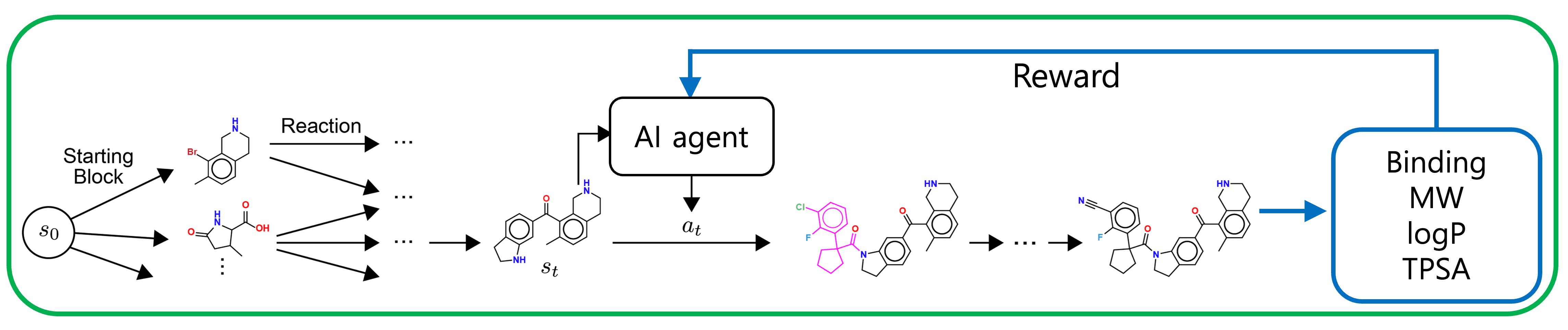
RxnFlow - 딥러닝으로 시간의 한계를 극복하다
RxnFlow를 알아보기 전, 가상 라이브러리의 원리부터 들여다 보겠습니다. 일반적으로 가상 라이브러리는 화학 반응물과 화학 반응의 조합으로 만들어집니다.
따라서 우리가 모든 가상 라이브러리를 계산하지 않고, 어떤 화학 반응물과 화학 반응의 조합이 타겟 Score를 올려줄 수 있는지 추론할 수만 있다면, 11조 개 분자를 모두 계산하지 않더라도 최적의 화학 반응물-화학 반응 조합을 찾을 수 있을 것입니다.
딥러닝은 바로 이걸 해내는 좋은 수단입니다. 데이터로부터 화학 반응물-화학 반응 조합과 타겟 Score의 관계를 파악하게끔 학습시키면 위와 같은 목적을 달성할 수 있습니다.
Target Score를 높이는데 특정 빌딩 블록들이 다른 빌딩 블록보다 중요할 것입니다. 이로부터 우리는 모든 빌딩 블록 조합을 같은 중요도로 탐색하는 것이 아니라, 특정 빌딩 블록 조합들을 더 집중적으로 탐색하는 것이 더 효율적이라는 것을 알 수 있습니다.
이러한 목적으로 GFlowNet이라는 모델이 적합합니다. 기존에는 강화학습이 이러한 목적으로 많이 사용되곤 했지만, 강화학습은 다양한 분자구조를 탐색하지 못하고 특정 분자 구조 주위만 탐색한다는 한계가 지적된 바 있습니다. 반면에 GFlowNet은 다양한 모드를 찾는데 강화학습보다 뛰어난 성능을 보여주기에 학계에 큰 주목을 받고 있습니다.
- GFlowNet이란?
- GFlowNet(Generative Flow Network, 생성 흐름 네트워크)은 강화학습(RL)과 생성 모델의 장점을 결합한 기법으로, 다양한 모드(Mode)를 가진 분포에서 샘플을 효율적으로 생성하는 것을 목표로 하는 기술입니다.
- GFlowNet은 특정 구조를 집중적으로 탐색하는 기존 강화학습과 달리, 넓은 화학 공간을 탐색하며 다양한 분자 구조를 생성할 수 있습니다. 또한, 주어진 목표(예: 높은 활성을 가진 분자)를 만족하는 다양한 구조를 빠르게 학습하여 분자를 생성할 수 있습니다. 이를 통해 신약 후보 물질을 효과적으로 발굴하고 신약 개발 프로세스를 가속화하는 데 기여할 수 있습니다.
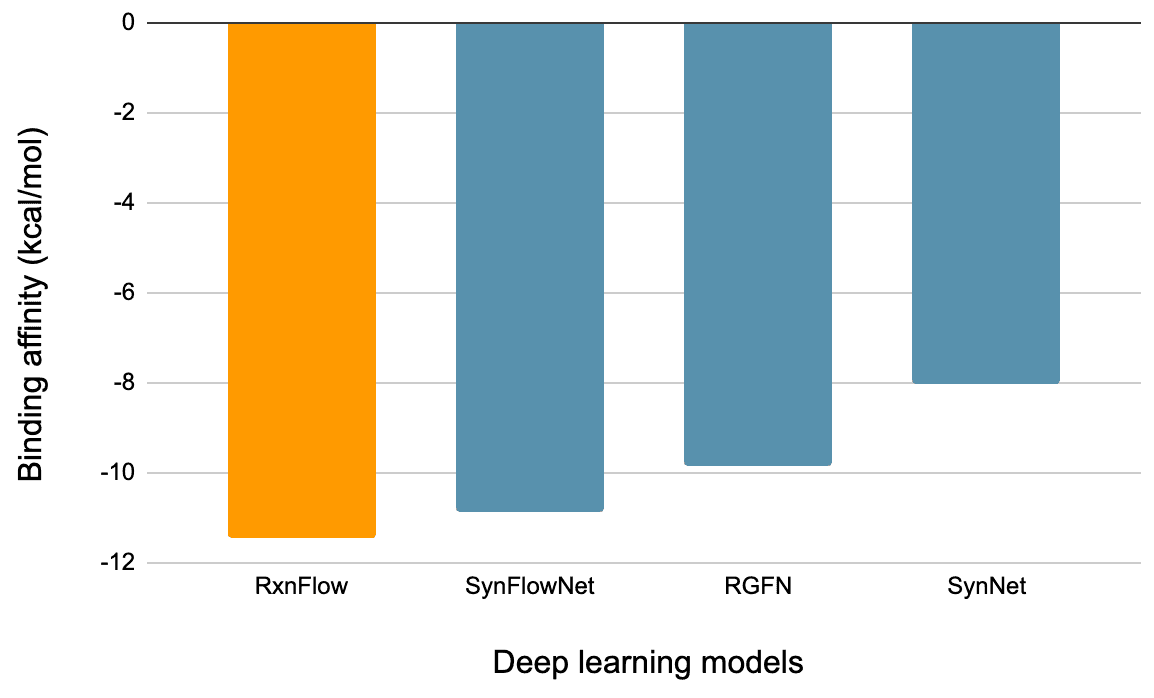
딥러닝을 활용한 결과
첫 번째 성능지표는 이렇게 찾은 분자의 결합 에너지입니다. 결합 에너지가 작으면 작을수록 AI모델이 더욱 우수한 물질을 찾은 것을 의미합니다. 아래 표에서 알 수 있듯이 RxnFlow로 생성한 분자가 다른 딥러닝 모델들보다 결합에너지가 작은 것을 알 수 있습니다.
다음은 합성 가능성입니다.
아래 표는 디자인한 전체 분자들 중 합성 가능한 분자의 비율을 의미합니다. 합성 가능 여부는 AiZynthFinder (Journal of Cheminformatics volume 12, Article number: 70 (2020)) 라는 합성 경로 예측 모델을 이용하여 판단하였습니다. 이 비율이 높으면 높을수록 딥러닝 모델이 합성 가능한 분자를 디자인하는 성능이 뛰어나다는 것을 의미합니다.
RxnFlow는 이 비율이 60%가 넘습니다. 지금까지는 AI로 생성한 분자가 합성 가능성이 낮다는 비판을 많이 받아왔으나, RxnFlow는 차원이 다른 우수한 성능을 보여주며 이러한 한계를 극복했습니다.

아래 그림은 RxnFlow로 찾은 물질과 합성경로를 시각화한 예시입니다. 연구자는 이 경로를 이용하여 직접 분자를 합성할 수도 있고, 분자 합성을 의뢰할 때 참고할 수도 있습니다. 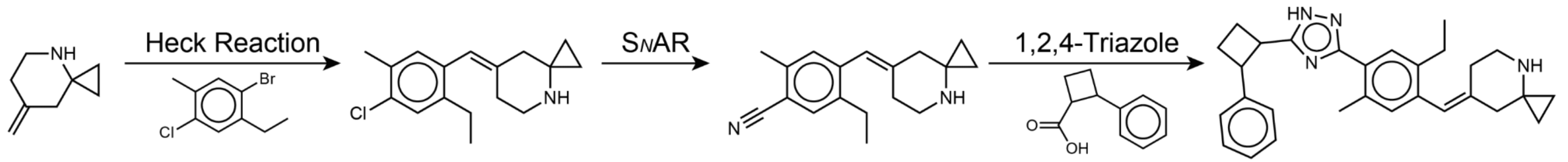
하이퍼랩(https://hyperlab.hits.ai/) 에서 새로 출시하는 기능인 Hyper Screening X는 이처럼 뛰어난 RxnFlow를 핵심기술로 하여 만들어졌습니다. 자신하건데, 초기 신약개발단계에서 빠른 시일내에 유효물질을 찾기 위한 최적의 방법은 Hyper Screening X를 이용하여 11조 개의 라이브러리를 가상 탐색하는 것입니다.
딥러닝이 탑재된 Hyper Screening X 출시일은?
현재 하이퍼랩에서는 Hyper Screening X 정식 출시를 준비 중이며, 곧 만나보실 수 있습니다. 하이퍼랩에 가입한 회원이라면, Hyper Screening X 정식 출시 완료 시점에 맞춰 이메일로 알려드리니 지금 먼저 회원가입을 완료해보세요. 회원가입 후 7일 동안 하이퍼랩 무료체험이 가능합니다.
AI 신약개발 플랫폼 하이퍼랩