최초의 AI-물리 융합 Docking, 신약 개발 패러다임을 바꾸다


안녕하세요, 히츠의 CTO 임재창입니다. 이번 글에서는 전통적인 분자 Docking의 한계를 넘어, 딥러닝과 물리 법칙을 융합하여 만든 PIGNet(Physics-Informed Graph Neural Network)의 개발 배경과 뛰어난 성능을 소개하고자 합니다.
AI와 물리학의 만남 : PIGNet의 혁신
리간드-단백질 결합 친화도 예측은 효과적인 약물 발견, 특히 초기 약물 개발 단계에서 히트 화합물을 식별하는 데 가장 중요한 기술 중 하나입니다. 딥러닝 기술이 등장하기 전 지난 30년 동안, 분자 Docking은 이 목적을 위해 가장 널리 사용된 방법론입니다. 이 글을 읽고 계시는 분들이라면, Schrödinger의 Glide, Discovery Studio의 GOLD, AutoDock Vina와 같은 유명한 Docking 소프트웨어에 익숙할 것입니다.
이러한 Docking 도구들이 존재했음에도 불구하고, 많은 실험 연구자들은 Docking의 낮은 정확도로 인해 단지 "보조 도구"로만 여겼습니다. 그러나 AI 기반 Docking 방법(줄여서 AI Docking)의 등장은 정확도에서 상당한 개선을 보여주며, 이러한 인식을 바꿀 수 있는 유망한 대안으로 주목받고 있습니다.
딥러닝 기술이 빠르게 발전하고 있음에도, 기존 AI Docking 방법의 근본적인 한계는 훈련 데이터가 부족한 영역에서 성능이 현저히 떨어진다는 점입니다. 이 문제를 해결하는 열쇠는 전통적인 Docking 방법에 있습니다. 해결책은 딥러닝과 전통 Docking 방법론의 강점을 결합하고 각각의 약점을 보완하는 데 있습니다.
이러한 아이디어는 PIGNet의 개발로 이어질 수 있었습니다. PIGNet은 리간드-단백질 상호작용 예측에서 높은 일반화 능력과 정확성을 동시에 달성했습니다. 이제 PIGNet이 이러한 성능을 어떻게 달성했는지 자세히 살펴보겠습니다.
물리 기반 방법론과 데이터 기반 방법론의 비교
분자 Docking과 분자 동역학 시뮬레이션은 물리 법칙을 기반으로 리간드-단백질 상호작용을 예측합니다. 이러한 방법은 수소 결합, 소수성 상호작용, 반데르발스 상호작용 등 연구자가 해석할 수 있는 다양한 원자 수준의 상호작용을 고려합니다. 물리 법칙은 보편적이기 때문에, 새로운 분자와 단백질 구조에 대해서도 정확성을 유지할 가능성이 높습니다. 또한, 특정 분자가 강한 결합 에너지를 갖는 이유를 해석할 수 있게 해줍니다.
그러나 물리 기반 방법의 단점은 계산 요구량에 있습니다. 정확한 물리 시뮬레이션은 계산 시간이 기하급수적으로 증가하며, 계산 비용을 줄이기 위해 근사치를 사용하면 정확도가 크게 떨어집니다. 그 결과, FEP+와 같은 비교적 정확한 방법은 상당한 자원과 시간을 필요로 합니다. 특히 이 한계점은 가상 스크리닝(virtual screening)에서는 큰 제약으로 작용합니다. 게다가 이러한 방법은 복잡한 초기 설정과 광범위한 사전 지식이 필요합니다. Docking과 같은 근사 방법을 사용해 해당 문제를 해결하면 계산 속도가 크게 향상되지만, 정확도는 상당히 낮아집니다.
반면, 딥러닝과 같은 데이터 기반 방법론은 데이터에서 직접 상관관계를 학습합니다. 명시적인 물리 법칙 없이, 리간드-단백질 결합 구조에서 필요한 특징을 추출하고 결합 에너지와의 관계를 학습합니다. 이 관계는 데이터에서 자동으로 학습되므로 물리적 근사를 필요로 하지 않아 근사 오류를 피하면서도, 빠른 속도를 보여줍니다.
하지만 데이터 기반 방법론에도 한계는 존재합니다. 충분한 데이터가 없을 때 성능이 저하될 가능성이 큽니다. 또한, 훈련 데이터와 유사한 분자에 대해서는 높은 성능을 보이지만, 그렇지 않은 분자에 대해서는 성능이 급격히 떨어집니다. 따라서, 이전에 보고되지 않은 새로운 화합물을 식별하는 것이 목표인 약물 발견의 경우에는 치명적인 약점으로 작용됩니다.
이처럼 두 방법론은 각각 뚜렷한 장점과 단점을 가지고 있습니다. 그렇다면 만약 두 방법론을 결합하여 서로의 장점을 극대화하고 단점을 상쇄할 수 있다면 어떨까요? 이러한 질문이 PIGNet(Physics-Informed Graph Neural Network)을 개발하게 된 동기가 되었습니다.
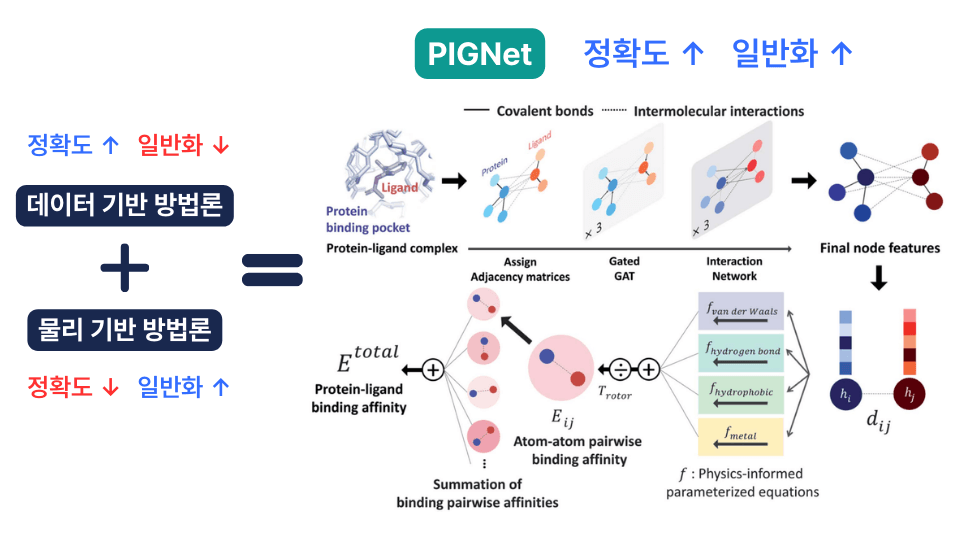
PIGNet: 물리 기반과 데이터 기반 방법론의 장점을 결합하다
PIGNet(물리 정보 그래프 신경망)은 딥러닝 방법론을 채택하여 물리 기반 근사의 한계를 극복하면서, 물리 법칙을 통해 데이터 기반 방법론의 과적합 문제를 완화합니다. PIGNet은 출력(결합 친화도)을 직접 예측하는 대신, 물리 방정식에 사용되는 매개변수를 예측합니다. 최종적으로 나오는 예측값은 딥러닝 모델이 예측한 매개변수와 물리 방정식을 결합해 산출됩니다.
먼저, 비결합 상호작용 중 하나인 반데르발스 에너지(van der Waals energy)는 방정식을 사용하여 계산됩니다. 이 방정식은 레너드-존스 포텐셜(Lennard-Jones potential)을 활용해 원자 간 거리에 기반한 상호작용 에너지를 계산합니다.
\[ E^{\text{vdW}} = \sum_{ij} c_{ij} \Bigg[\Bigl(\frac{d_{ij}'}{d_{ij}}\Bigr)^{12} - 2 \Bigl(\frac{d_{ij}'}{d_{ij}}\Bigr)^6\Bigg] \]
수소 결합, 소수성 상호작용, 금속-리간드 상호작용은 또 다른 방정식을 통해 계산됩니다.
\[ e_{ij} =
\begin{cases}
w, & \text{if } d_{ij} - d_{ij}' < c_1,\\
w \Bigl(\frac{d_{ij} - d_{ij}' - c_2}{c_1 - c_2}\Bigr), & \text{if } c_1 < d_{ij} - d_{ij}' < c_2,\\
0, & \text{if } d_{ij} - d_{ij}' > c_2,
\end{cases} \]
마지막으로, 모든 상호작용 에너지를 합산하고 분자의 회전 자유도를 고려한 정규화 계수로 나누어 총 에너지를 계산합니다.
\[ E^{\text{total}} = \frac{E^{\text{vdw}} + E^{\text{hbond}} + E^{\text{metal}} + E^{\text{hydrophobic}}}{T_{\text{rotor}}}. \]
딥러닝 모델은 최종 예측의 정확도를 최적화하도록 훈련되며, 훈련 후에는 추론 과정에서 모델이 이러한 매개변수를 예측합니다. 다음 섹션에서는 PIGNet의 성능을 유도체의 결합 친화도 예측과 가상 스크리닝이라는 두 가지 관점에서 살펴보겠습니다.
성능 1: 유도체 결합 친화도 예측
유도체 활성 예측 정확도는 다양한 유도체의 활성을 얼마나 정확하게 예측할 수 있는지를 측정합니다. 이 정확도는 실험 값(IC50, Ki 등)과 예측 값 간의 피어슨 상관계수(Pearson correlation)를 통해 정량화됩니다. 이 값이 1에 가까울수록 예측이 정확합니다. 아래 그래프에서 보듯, PIGNet은 다른 주요 도킹 방법론에 비해 두 배 이상의 정확도를 보여줍니다.
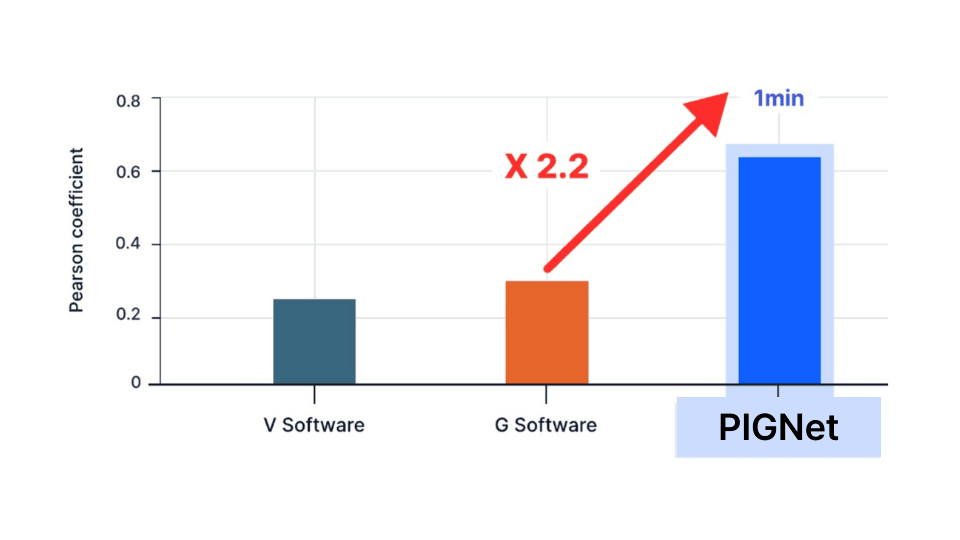
참고 문헌: Digital Discovery, 2024, 3, 287-299벤치마크 세트: J. Chem. Inf. Model., 2020, 60, 5457–5474
성능 2: 가상 스크리닝
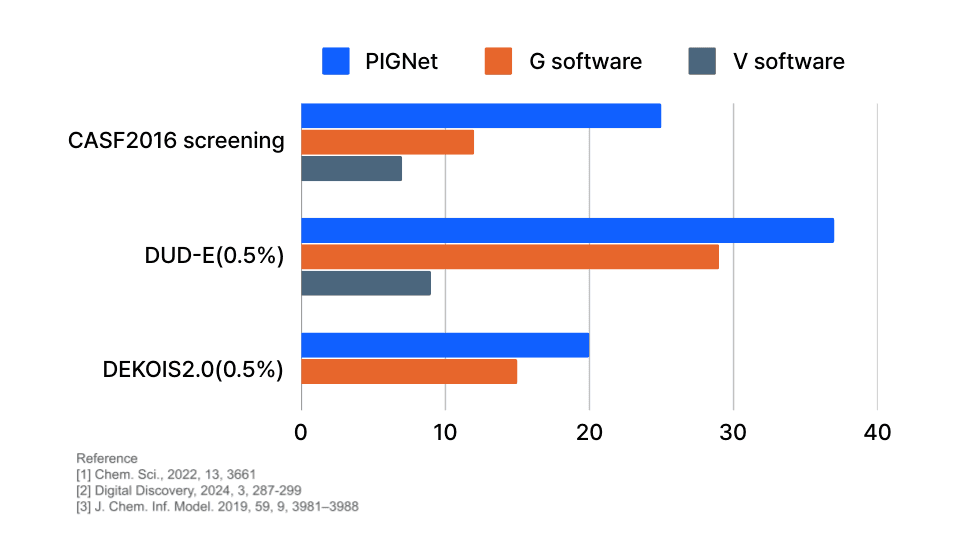
가상 스크리닝의 성능은 농축 인자(Enrichment Factor, EF)로 측정할 수 있습니다. 아래 그래프에서 확인할 수 있듯이, PIGNet은 여러 데이터셋에서 기존 도킹 방법보다 최대 두 배 높은 EF를 보여줍니다. 이는 가상 스크리닝에서 활성 화합물을 찾아낼 확률이 두 배 더 높다는 뜻입니다.
Hyper Lab에서 PIGNet 무료 체험하기
지금까지 빠르게 발전하는 딥러닝 기술과 오랜 시간 축적된 물리 지식을 융합한 PIGNet의 이론적 배경과 성능 결과를 살펴봤습니다. PIGNet은 현재까지 달성된 최고 수준의 정확도를 자랑하며, 약물 발견에서 제한된 데이터 문제를 효과적으로 해결하고 있습니다. 이 기술은 Hyper Lab의 Hyper Binding 기능의 핵심 모듈로 활용되고 있으며, Hyper Lab에서는 가장 정확한 리간드-단백질 상호작용 예측 모델을 무료로 체험해 보실 수 있습니다. 홈페이지 상단의 ‘지금 무료 체험하기’ 버튼을 클릭하시면 간단한 회원가입으로 바로 시작하실 수 있습니다.
AI 신약개발 플랫폼 하이퍼랩