ICLR 2025 미리 보기


ICLR이란?
안녕하세요. 히츠의 AI 연구 1팀의 연구원 배성한 입니다. 이번 4월 싱가포르 엑스포에서 인공 지능 연구자들이라면 누구나 주목할 만한 이벤트가 열리는데요, 바로 2025 ICLR입니다. ICLR(International Conference on Learning Representations)은 OpenReview 플랫폼을 통해 투명성을 높이고, 인공 지능 연구자 간의 활발한 토론을 촉진하고자 만들어진 인공지능 국제 학회입니다. 지난 2013년 딥러닝 선구자인 Yoshua Bengio와 Yann LeCun 등의 주도로 처음 개최됐는데요. ICLR은 매년 영향력 있는 머신러닝 및 인공지능 분야 연구들이 발표되어 지난 포스트에서 소개된 ICML, NeurIPS와 함께 전 세계 인공지능 분야 연구자와 종사자들이 가장 주목하는 학회 중 하나입니다.
최근 인공 지능 기반 신약 개발이 많은 주목을 받는 만큼 이번 2025 ICLR에서도 다양한 관련 연구들이 소개될 예정인데요. 이번 포스트에서는 이들 중 인상적인 연구 몇 가지를 간략하게 소개하고자 합니다.
ICLR 2025 미리 보기: Generative Flows on Synthetic Pathway for Drug Design
분자 생성 모델은 최근 구조 기반 약물 설계(Structure-Based Drug Design; SBDD) 에서 가장 주목받는 기술들 중 하나입니다. 이미지, 자연어 분야에서 큰 성공을 거둔 생성 모델을 신약 설계 분야에 적용하여 특정 타깃 단백질에 대해 효과적일 것이라고 여겨지는 약물 분자(리간드)를 AI 모델이 생성하는 것이죠.
이러한 인공지능 기반 약물 분자 생성은 많은 계산량과 시간이 소모되는 전통적 가상 스크리닝 방법을 대체할 꿈의 기술로 주목받고 있습니다. 하지만 기존의 분자 생성 모델들은 대개 ‘합성 가능성’을 고려하지 않기 때문에 좋은 물성을 가질 것으로 예상되더라도 실제 물질을 만들기가 어려운 경우가 많았습니다.
이번에 소개해드릴 논문은 Generative Flows on Synthetic Pathway for Drug Design입니다. 이 논문에서는 정의된 분자 빌딩 블록과 화학 반응 템플릿을 사용하여 분자를 차례대로 조합하는 방식을 사용하여, 합성 가능성이 높은 리간드들을 생성하는 AI 모델인 RxnFlow를 제시하였습니다.
핵심 방법론 설명
-
순차적 분자 생성 프로세스
RxnFlow는 GFlowNets 구조를 차용하여 순차적 생성 프로세스를 통해 효과적으로 학습 가능한 분자를 생성합니다. GFlowNets은 지난 2021년에 발표된 GFlowNet Foundations 논문에서 소개된 AI 기법으로, 초기 상태(initial state) \($s_0$\)에서 최종 상태(terminal state) \($s_f$\)로 가는 흐름(flow) \($F(s\to s')$\)를 학습합니다. 이때 각 state로 들어오는 flow의 합과 나가는 flow합은 동일하며, 이는 각 state로 도달할 확률과 비례합니다. 또한 terminal state에 도달하는 flow는 우리가 목적으로 하는 상태를 표현하는 보상(reward) \($R(s_f)$\)과 비례합니다.
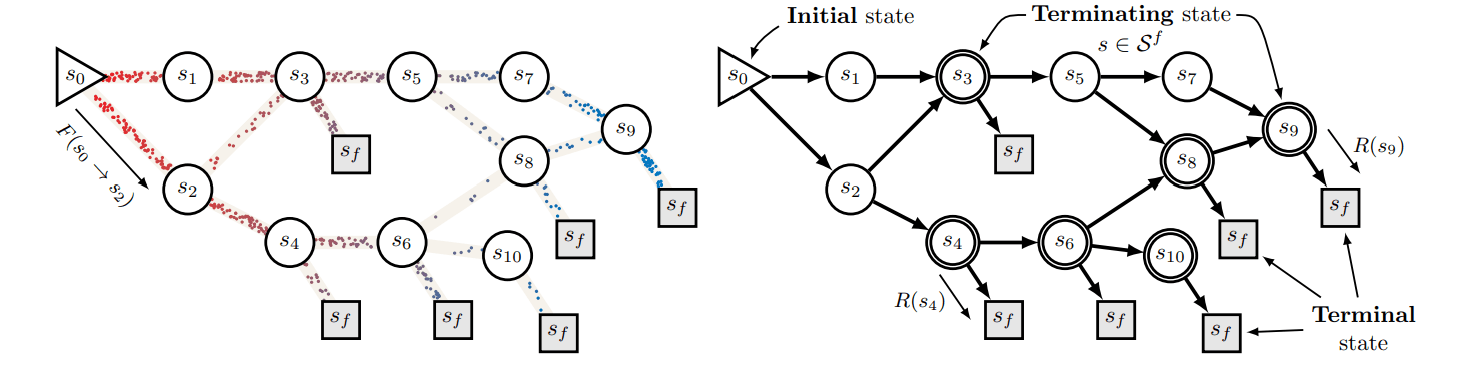
그림 1. GFlowNets 모식도. [출처] BENGIO, Yoshua, et al. Gflownet foundations. The Journal of Machine Learning Research, 2023, 24.1: 10006-10060. 리간드를 생성하는 RxnFlow에서 initial state는 분자를 생성하기 위한 빌딩 블록 (building block), terminal state는 후보 리간드 분자, 그리고 flow는 빌딩 블록에서 리간드를 합성하기 위한 합성 경로로 정의됩니다. 이때 합성 경로는 실제 존재하는 빌딩 블록과 구현 가능한 반응 템플릿을 통해 정의되기 때문에 해당 flow는 높은 합성 가능성을 보장합니다.
따라서 특정 단백질에 대해 원하는 리간드의 특성(예: 특정 단백질 타겟에 대한 결합력)을 보상으로 설정하면, RxnFlow는 해당 특성을 만족하면서 합성 가능성이 높은 다양한 후보 리간드 분자를 생성할 수 있습니다.
-
Action Space 서브 샘플링
RxnFlow에서 Action Space는 120만 개의 빌딩 블록과 71개의 반응 템플릿을 조합함으로써 구성됩니다. 이러한 빌딩 블록과 반응 템플릿의 조합을 통해 합성이 가능한 분자를 생성해줍니다. 그러나 여전히 학습 과정에서 모두 고려하기에는 너무 큽니다. 이러한 방대한 Action Space를 효과적으로 학습시키기 위해서 RxnFlow에서는 Action Space 서브 샘플링 방법을 구현하였습니다.
아래 그림 2를 보면, RxnFlow는 빌딩 블록과 반응 템플릿을 신경망으로 임베딩(embedding)한 뒤, 중요도 기반 서브 샘플링을 통해 Action Space를 효과적으로 축소합니다. 전체 학습 과정에서는 축소된 Action Space 내에서 현재 state에서 다음 state로 이동하는 action을 고르면서 결과적으로 terminal state가 보상을 최대화하는 방향으로 학습됩니다. 이때 서브 샘플링된 Action Space에 가중치를 두어 제한된 Action Space를 보완해 줍니다.
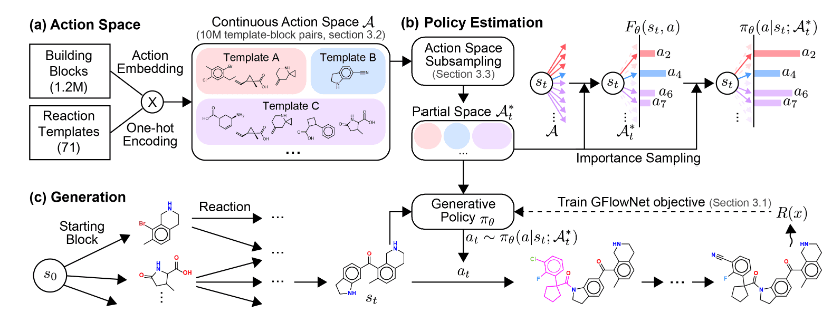
그림 2. Action Space 서브 샘플링을 통한 RxnFlow 학습 과정. [출처] SEO, Seonghwan, et al. Generative flows on synthetic pathway for drug design. arXiv preprint arXiv:2410.04542, 2024. - 비계층적 MDP
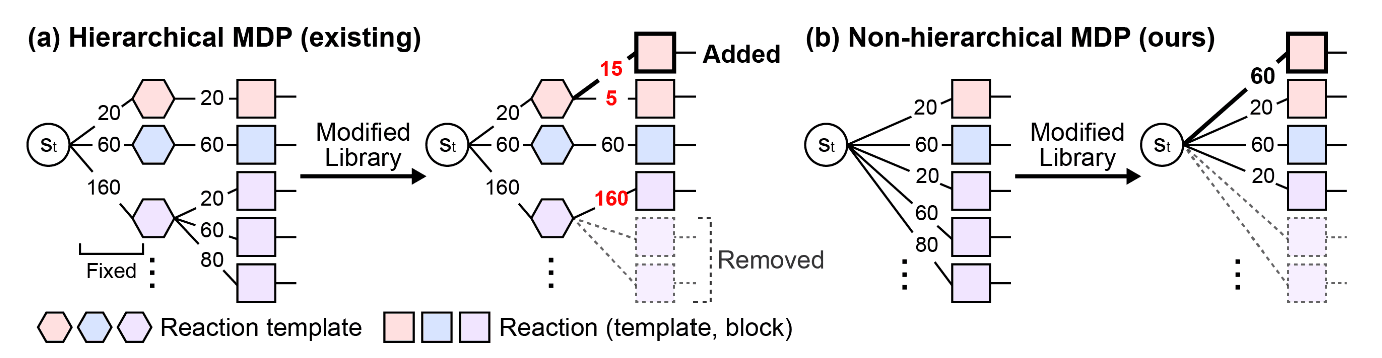
기존의 합성 기반 분자 생성 모델들은 합성 경로를 선택할 때, 먼저 반응 템플릿에 대한 flow를 추정합니다. 그다음, 해당 반응 템플릿 내에서 종속된 빌딩 블록으로 flow가 흐르는 방식인 계층적 MDP(Markov Decision Process)를 따릅니다. RxnFlow의 저자들은 이러한 계층적 MDP 방법이 아래 그림 3.(a) 처럼 일단 반응 템플릿이 정해지면 제한된 빌딩 블록으로만 선택할 수 있어 결과적으로 잘못된 flow로 유도되는 한계를 지적합니다.
RxnFlow는 이러한 한계를 극복하기 위해 아래 그림 3.(b) 처럼 반응 템플릿과 빌딩 블록을 동시에 선택하는 비계층적 MDP 방법을 제시하여 더 다양한 리간드 생성 flow를 안정적으로 학습시킬 수 있었습니다.
결과
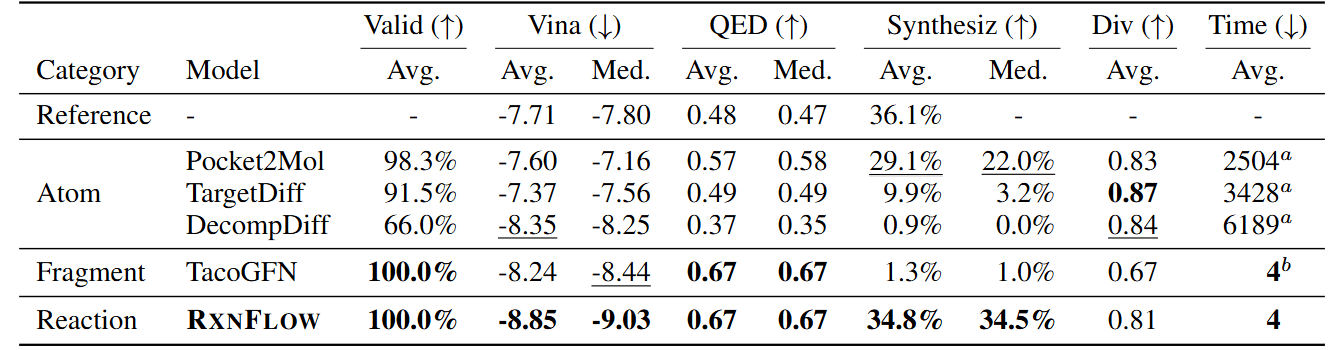
앞서 설명한 방법들을 통해 RxnFlow는 여러 타겟 포켓에 대해 실험적으로 기존의 반응 기반 분자 생성 모델보다 더 나은 성능을 보였습니다. CrossDocked2020 벤치마크 내 단백질들을 대상으로 RxnFlow는 평균 Autodock Vina score -8.85 kcal/mol의 리간드들을 34.8%의 합성 가능성을 보고했습니다. 이를 통해 RxnFlow는 기존의 분자 생성 모델들에 비해 더 효과적인 약물을 효율적으로 생성하는 동시에, 생성해낸 분자들의 신뢰성이 제한적이라는 기존의 분자 생성 모델의 한계를 극복해냈다는 것을 입증했습니다.
ICLR 2025 미리 보기: Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows
SBDD(Structure-Based Drug Design)에서 단백질의 동적 특성은 중요한 고려 대상 중 하나입니다. 단백질의 동적 특성이란 단백질이 리간드와 결합할 때 결합한 리간드와의 상호작용을 최적화하기 위해 구조적으로 변화하는 특성을 말하는데요. 이를 유도 적합(induced fit)이라고 합니다. 이때 단백질의 결합 전 구조를 아포(apo) 구조, 그리고 리간드와 결합 뒤 변화한 구조를 홀로(holo) 구조라고 합니다.
대부분의 기존 SBDD 접근 방식은 계산의 효율화를 위해 단백질의 구조를 정적이라고 가정하여 실제 적합한 약물 분자의 발견에 한계가 있었습니다. 분자 동역학(Molecular Dynamics; MD) 시뮬레이션 기법은 단백질 분자의 동적인 변화를 포착할 수 있습니다. 하지만 많은 계산 시간과 비용을 요구하여 대규모 신약 개발 프로세스에 적용하기에는 한계가 있습니다.
이러한 한계점들을 극복하기 위해 본 논문에서는 MD 시뮬레이션을 통해 아포 및 여러 홀로 상태의 단백질-분자 복합체 데이터를 수집하고, 단백질 내에 리간드가 결합하는 부위인 포켓(pocket)의 변형을 학습하는 인공지능 모델인 DynamicFlow를 제안합니다.
핵심 방법론
-
MD 기반 apo-holo 데이터 생성
제한된 단백질-리간드 데이터셋은 단백질의 동적 특성을 반영하는 인공지능 모델의 학습에 가장 큰 병목이었습니다. 저자들은 이러한 데이터의 한계를 극복하기 위해 약 20,000개 단백질-리간드 복합체의 MD 시뮬레이션 데이터를 포함하고 있는 MISATO 데이터셋을 활용하였습니다.
해당 데이터셋 내에 각 MD 시뮬레이션 궤적에 따라 단백질 포켓을 설정하여 holo 구조를 정의하고, MD 시뮬레이션 궤적 내의 리간드의 RMSD 기준으로 클러스터링하여 유사한 holo 구조들 사이에 대표 구조를 선정하여 훈련 데이터에 사용하였습니다. apo 구조는 해당 단백질의 서열을 기반으로 AlphaFold2가 예측한 구조를 사용함으로써 결과적으로 저자들은 총 5,692개의 apo, holo 구조 및 리간드 상호작용 데이터 셋을 구축하였습니다.
-
다중 스케일 full-atom 네트워크
DynamicFlow 모델은 타깃 단백질의 apo 상태를 holo 상태로 변환하면서, 동시에 holo 구조에 도킹된 리간드를 생성합니다. 이때 입력으로 들어오는 단백질의 apo 구조 및 노이즈가 추가된 리간드를 원자 단위로 쪼개서 임베딩(embedding)합니다. 이를 다중 스케일 full-atom 네트워크라 하고 전체 구조는 다음과 같은 두 가지 구성 요소로 이루어져 있습니다.
-
원자 레벨 SE(3) 등가 그래프 신경망(GNN)
먼저 apo 단백질 및 노이즈가 추가된 리간드 분자를 각 원자를 노드로 하는 그래프로 구축합니다. 이후 그래프 신경망(GNN)을 통해 그래프 내 각 원자들을 회전 및 평행이동(SE(3) 변환)에 등가적으로 임베딩 합니다. 해당 네트워크에서 나온 리간드의 원자의 hidden state는 이후 노이즈를 제거하며 리간드 내 원자와 결합 유형, 그리고 단백질 holo구조 내 도킹 조를 생성해내는 데 사용됩니다.
-
residue 레벨 트랜스포머(Transformer)
위의 GNN에서 나온 단백질 원자의 hidden state는 각 속해진 잔기(residue) 수준으로 합쳐집니다. 그리고 residue 프레임의 좌표와 뒤틀림 각(torsion angle)과 함께 input으로 구성한 뒤, residue 레벨 Transformer 네트워크를 통해 residue 프레임의 3차원 변환 및 뒤틀림 각을 업데이트하여 holo 구조를 예측합니다.
-
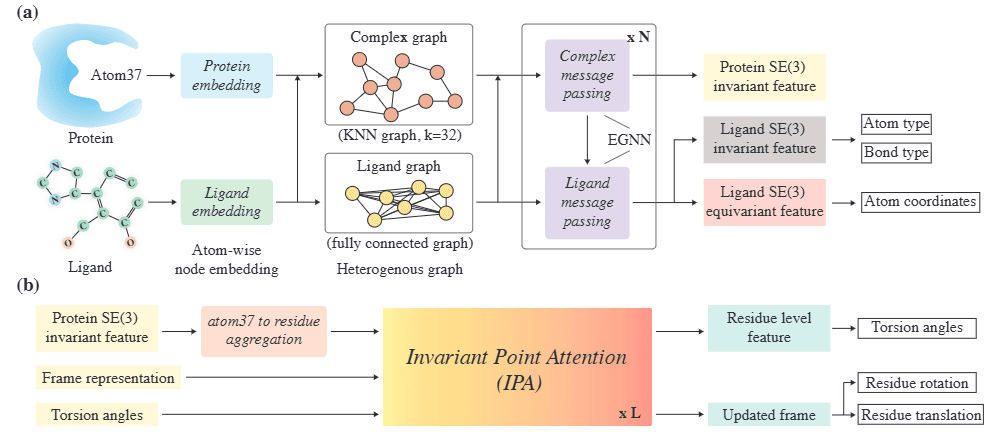
-
Flow matching을 통한 단백질-리간드 동적 시스템 학습
앞서 설명 드린 다중 스케일 full-atom 네트워크는 flow matching을 통해 학습 됩니다. Flow matching이란 단순한 초기 상태의 분포 \($p_0$\) 를 우리가 목표로 하는 데이터의 분포 \($p_1$\)를 변환하는 경로를 직접 학습시키는 생성 모델 기법입니다.
이때 모델은 초기 상태에서 목표 상태로 가는 경로를 미분한 벡터 필드(flow)를 맞추도록 학습됩니다. 추론 시에는 모델이 현재 상태를 기반으로 flow를 추론하고, 이를 기반으로 상미분 방정식 (Ordinary Differential Equation; ODE)을 풀어 상태를 업데이트하며 차례대로 목표 상태에 도달하는 방식입니다.
DynamicFlow에서는 단백질의 apo 구조 및 노이즈가 추가된 리간드를 초기 상태, holo 구조 및 도킹 리간드 상태가 목표 상태로 설정합니다. 그리고 이 두 상태를 보간하여 중간 상태들을 미리 정의한 뒤, 상태 별 flow를 맞추도록 full-atom 네트워크를 학습시킵니다. 추론 시에는 해당 네트워크가 추정한 flow를 따라 ODE를 풀며 apo구조를 holo구조로 변환하는 동시에 도킹 리간드를 생성해냅니다.
또한 저자들은 학습 과정에서 생성해 낸 중간 상태들에 가우시안 노이즈를 첨가하여 학습한 DynamicFlow-SDE (Stochastic Differential Equation)를 도입하여, 높은 차원의 노이즈에 대해 더욱 강건한 모델을 만들었습니다.
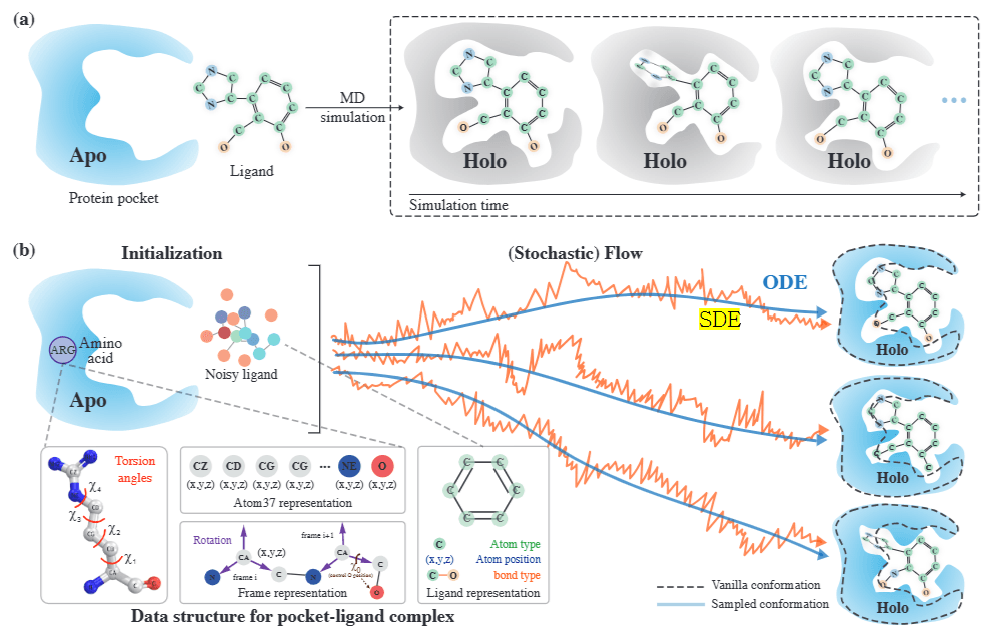
결과
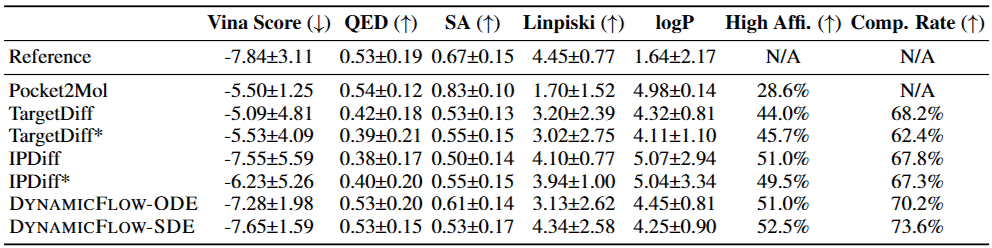
DynamicFlow의 저자들은 MISATO 중 일부를 가져와 특정 타깃에 대한 리간드 생성 성능을 평가하였습니다. 이때 DynamicFlow는 정적 단백질 구조를 사용하는 기존 SBDD 모델보다 종합적으로 더 우수한 리간드를 생성했습니다.
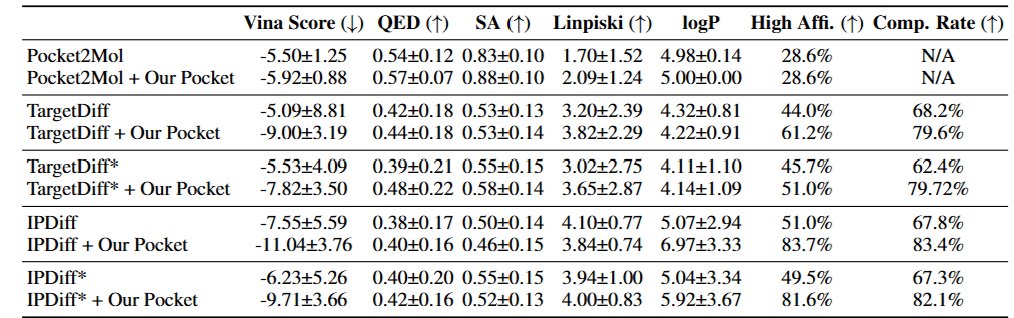
또한, 기존 SBDD에 DynamicFlow의 holo 구조를 입력하면 더 우수한 리간드를 생성함을 실험적으로 증명했습니다. 이처럼 DynamicFlow는 단백질의 동적 특성을 활용하여 더욱 효과적으로 유망한 리간드를 발견하고, 기존 모델보다 향상된 holo 예측 구조를 제공하여 구조 기반 약물 개발 분야에 혁신적인 기여할 것으로 기대됩니다.
ICLR 2025 미리 보기: NEXT-MOL: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation
이번에 소개해드릴 논문은 1 차원 형태의 분자를 생성하는 언어 모델을 통해 3차원 분자 생성 기능을 개선한 NEXT-MOL 모델을 소개합니다. 기존 분자 생성 연구들은 주로 3차원 확산 모델을 활용하여 분자 구조 생성의 성능을 개선해 왔습니다. 하지만 본 논문의 저자들은 항상 유효한 분자를 생성하면서도 수십 억 개 규모의 1차원 분자 데이터셋을 활용할 수 있는 SELFIES 기반 언어 모델(LM)의 장점이 그동안 간과되어 왔다고 지적합니다.
NEXT-MOL은 3D 분자를 생성하는 기초 모델입니다. 먼저 분자 생성 언어 모델을 사용하여 1차원 분자 서열(SELFIES)을 생성하고, 이후 확산 모델을 사용하여 해당 서열의 3차원 구조(conformer)를 예측하는 두 가지 단계의 방법을 사용하여 리간드 생성 성능을 향상 시켰습니다.
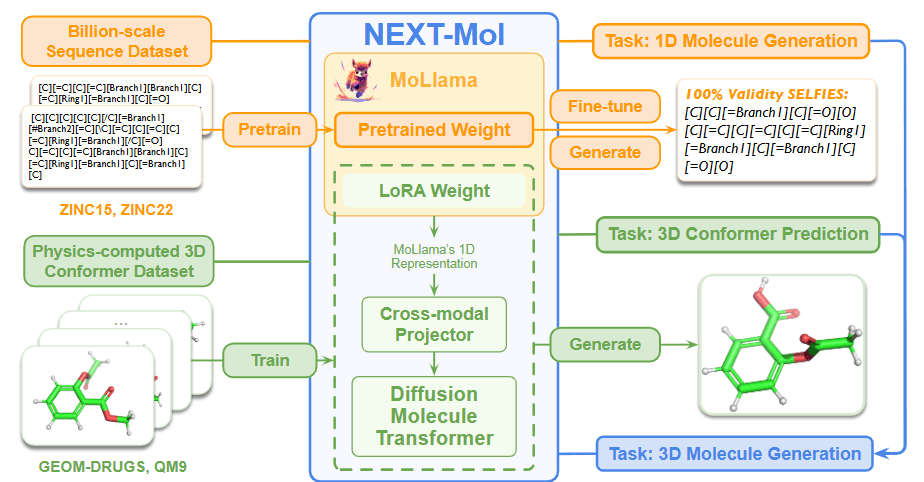
핵심 방법론 설명
-
1 차원 SELFIES 생성 언어 모델(MoLlama)
MoLlama는 960M 파라미터를 가진 언어 모델로, 다음 토큰 예측을 통해 1 차원 분자 서열을 생성합니다. 해당 논문의 저자들은 ZINC-15 데이터베이스에서 수집된 1.8억 개의 분자들을 SELFIES로 변환 및 정제하여 구성한 훈련 데이터를 통해 MoLlama를 사전 학습시켰습니다. 또한 같은 분자라도 2차원 분자 그래프의 순서를 무작위로 바꿈에 따라 다른 SELFIES가 생성됨에 착안하여, 무작위 SELFIES 데이터 증강을 통해 fine-tuning 시킵니다.
이를 통해 MoLlama는 분자 내 원자의 순서에 구애받지 않게 됩니다. 결과적으로 모델의 과적합(over-fitting)이 완화되고 학습 과정에서 보게 되는 분자의 본질적 다양성이 향상됩니다. 이렇게 광범위한 사전 학습 및 fine-tuning을 거친 MoLlama는 scaffold나 fragment 같은 이후 3차원 conformer 예측에 유용한 분자 패턴에 대한 정보를 제공합니다.
-
3D 구조 예측 모델(DMT)
Diffusion Molecular Transformer(DMT)는 앞선 MoLlama가 생성해 낸 1차원 SELFIES 서열의 3차원 conformer를 예측하는 데 사용되는 모델입니다. DMT는 RMHA(Relational Multi-Head Self-Attention) 네트워크를 활용하여 구성되었습니다. 해당 아키텍처는 분자 내 각 원자의 표현과 원자 쌍 간의 관계 표현을 동시에 업데이트하며 전체 분자에 대해 효과적으로 임베딩합니다.
DMT는 diffusion을 통해 Diffusion이란 원본 데이터 \($x_0$\)에 연속적으로 노이즈를 추가하여 단순한 노이즈 상태의 데이터 \($x_T$\)로 변환한 뒤, 이 역과정을 신경망이 학습하는 생성 모델 기법입니다. 추론 시에는 네트워크가 노이즈가 첨가된 입력을 받아 점진적으로 노이즈를 제거하면서 최종적으로 우리가 원하는 특성의 데이터를 생성할 수 있게 됩니다. DMT 역시 3차원 conformer 정보를 갖고 있는 GEOM-DRUGS나 QM9 데이터셋을 통해 사전 학습되며, 아래 그림 6. (a)처럼 노이즈가 추가된 분자를 입력으로 받아 추가된 노이즈를 예측함으로써 원본 분자를 복원하도록 학습됩니다.
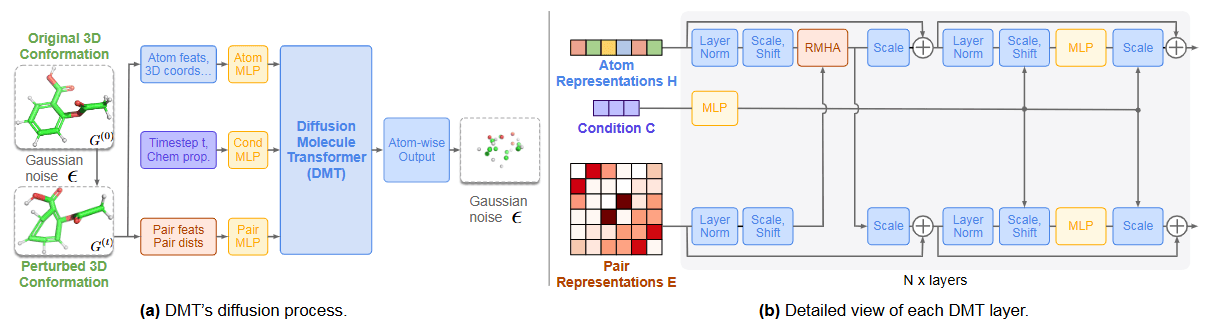
그림 6. Diffusion Molecular Transformer(DMT) 네트워크의 구조. [출처] Anonymous authors. NEXT-MOL: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation. OpenReview. -
전이 학습 (Transfer Learning)
NEXT-MOL는 사전 학습된 MoLlama에서 생성한 1차원 SELFIES 정보를 역시 사전 학습된 DMT에 넘겨주는 전이 학습을 통해 전체 3차원 리간드 생성 성능을 개선했습니다. 전이 학습 과정 동안 타깃 분자는 MoLlama를 통해 형성된 1차원 임베딩 및 2차원 분자 그래프를 통해 DMT의 입력으로 변환됩니다. 이때 MoLlama로 나온 1차원 임베딩을 DMT가 이해할 수 있도록 변환하는 역할을 하는 cross-modal projector 네트워크가 도입되어 같이 학습됩니다.
전체적인 전이학습 과정은 아래 그림 7. (b)와 같습니다. 먼저 앞서 언급한 것처럼 3차원 conformer 정보를 가진 데이터들로 DMT를 사전 학습시킨 뒤, 사전 학습 된 MoLlama와 DMT를 cross-modal projector로 연결합니다. 이후 DMT의 파라미터를 고정한 상태에서, MoLlama의 LoRA 네트워크와 cross-modal projector를 짧은 에폭(Epoch) 동안 학습시킵니다. 저자들은 이 과정이 새롭게 도입된 cross-modal projector의 무작위 파라미터가 사전 학습된 DMT 내의 파라미터가 왜곡된 방향으로 유도되는 것을 방지하는 역할이며, 이를 projector warmup이라고 언급하였습니다. projector warmup이 끝난 NEXT-MOL은 MoLlama(내의 LoRA)와 cross-modal projector, DMT를 모두 fine-tuning하며 전이 학습을 마무리 합니다.
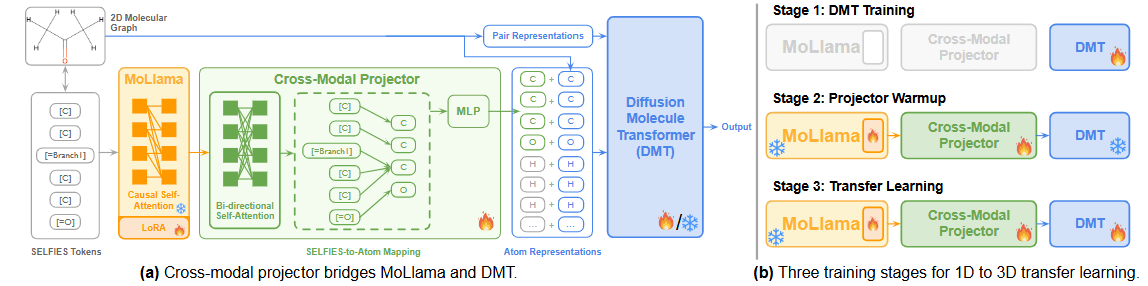
그림 7. cross-modal projector 구조와 전이 학습 과정 [출처] Anonymous authors. NEXT-MOL: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation. OpenReview.
결과
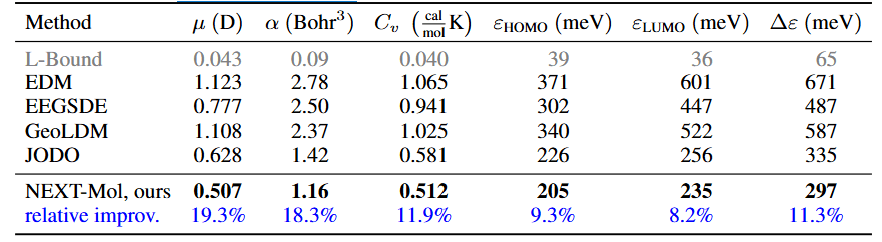
사전 학습 및 전이 학습이 끝난 NEXT-MOL은 MoLlama를 통해 먼저 1차원 SELFIES를 생성한 뒤, 이를 기반으로 DMT에서 conformer를 예측하여 최종적으로 3차원 리간드를 생성합니다. NEXT-MOL은 다양한 실험을 통해 DMT 모델의 3차원 분자 생성 및 구조 예측 성능의 향상을 입증하였습니다. 1차원 분자 생성 언어 모델의 활용을 통해 더 폭 넓은 분자 데이터 활용할 수 있으며, 새로운 화합물 생성에 유용한 기초 모델로서의 가능성을 보여주었습니다.
ICLR 2025: AI가 창조하는 신약 개발의 미래
오늘은 2025 ICLR에 새롭게 발표되는 인공 지능 신약 개발 연구 중 인상적인 몇 개에 대해 알아보았습니다. 해당 연구들을 통해 우리는 최근 인공 지능 기반 신약 개발 연구의 주요한 트렌드 중 하나가 Diffusion 혹은 Flow matching과 같은 생성 모델 기법을 기반으로 타깃 단백질에 효과적으로 결합하는 약물 분자를 생성해 내는 구조 기반 분자 생성임을 알 수 있었습니다.
또한, 생성된 분자의 합성 가능성을 높이기 위해 다양한 접근법이 적용되었습니다. RxnFlow는 빌딩 블록과 반응 템플릿을 활용해 순차적 분자 합성 프로세스를 학습하였으며, DynamicFlow는 MD 시뮬레이션을 이용해 단백질-리간드 결합의 induced fit을 고려했습니다. NEXT-MOL은 부족한 3차원 conformer 데이터를 보완하기 위해 대규모 1차원 분자 서열 데이터로 사전 학습한 언어 모델을 활용했습니다. 이를 통해 AI 기반 분자 생성의 품질이 비약적으로 향상되었습니다.
이번 2025 ICLR에서 살펴보면서 AI가 약물 분자의 특성이나 단백질의 구조를 “예측”하는 능력을 넘어서서 이제 새로운 약물을 “창조”하는 영역에 접어들게 되었음을 알 수 있었습니다. 즉, 이제 AI가 주도하는 신약 개발의 시대는 이는 거부할 수 없는 흐름이 되었다고 해도 과언이 아닙니다. 앞으로 AI가 신약 개발에 어떠한 혁신을 불러올지, 그리고 이로 인해 전 인류의 건강과 수명에 어떠한 영향을 끼칠까요? 앞으로의 미래가 기대됩니다.
이러한 변화 속에서 Hyper Lab은 AI 기반 신약 개발을 더욱 효율적으로 지원하는 역할을 하고 있습니다. 자체 개발한 AI를 활용해 초기 신약 후보 물질을 빠르게 탐색할 수 있으며, 연구자들이 쉽게 활용할 수 있도록 설계되었습니다. 무료 체험도 가능하니, 지금 바로 Hyper Lab을 통해 AI 신약 개발의 가능성을 직접 경험해 보시길 추천합니다.
AI 신약개발 플랫폼 하이퍼랩