NeurIPS 2024 미리 보기


NeurIPS 2024
NeurIPS(Neural Information Processing Systems)는 인공지능, 기계학습, 딥러닝 등 신경망 및 정보 처리 시스템 관련 연구자들이 발표하고 교류하는 대표적인 학회입니다. 1986년 미국에서 시작된 이 학회는 초기에는 생물학적 신경망과 인공 신경망 모두를 다루었지만, 현재는 주로 인공 신경망을 중심으로 한 딥러닝 및 기계학습 연구가 주를 이루고 있습니다.
NeurIPS는 AI 연구의 최전선에 있는 학회로, 매년 최신 기술과 연구 결과가 발표되는 자리입니다. 특히 컴퓨터 비전, 자연어 처리, 강화 학습, 생성 모델 등 다양한 분야에서 중요한 연구가 소개됩니다. 최근에는 AI for Science가 중요한 연구 주제로 떠오르면서, 신약 개발이나 생물학적 연구와 같은 과학 분야에 AI를 응용하는 논문도 증가하고 있습니다.
NeurIPS는 논문 발표뿐만 아니라 워크숍, 튜토리얼, 포스터 세션, 챌린지 등을 통해 연구자들이 실질적으로 기술을 익히고 협력할 수 있는 기회를 제공하여, AI와 딥러닝 커뮤니티에서 가장 영향력 있는 학회로 자리 잡고 있습니다.
이번 포스팅에서는 올해 12월, 밴쿠버에서 개최되는 NeurIPS 학회에 대한 정보를 소개해 드리겠습니다.
NeurIPS 2024 에 발표되는 신약개발 관련 분야 연구들
승인 논문 목록에서 molecule, drug, protein 등의 키워드로 신약개발 관련한 연구들을 모아보았습니다.
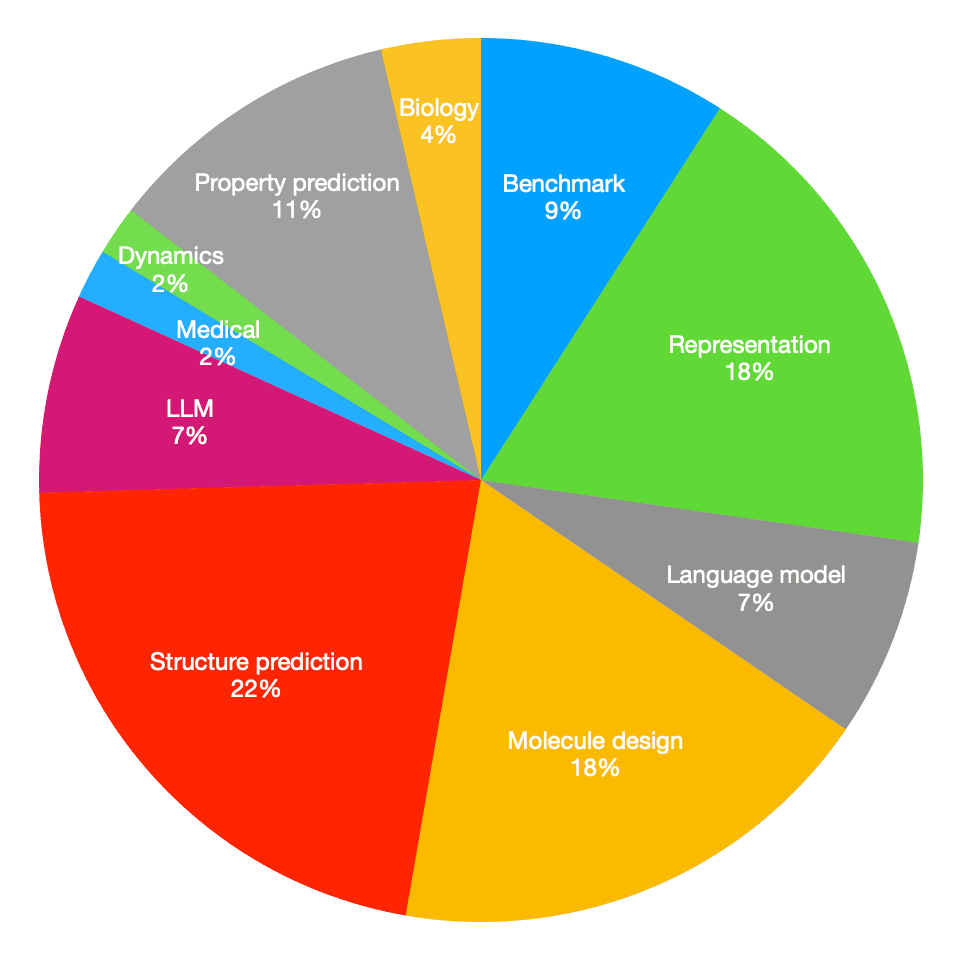
올해 다뤘던 ICLR 2024, ICML 2024와 마찬가지로 molecule design과 representation에 대한 연구가 많았습니다. 특히 분자를 디자인할 때 타겟 단백질의 포켓 구조를 고려하면서, 특정 조건(합성 가능성, 물성 등)을 동시에 만족시키는 분자 설계 방식이 눈에 띄게 많았습니다.
한편, 지난 두 학회와는 다르게 구조 예측과 벤치마크를 정립한 연구도 많았는데요. 벤치마크 연구는 기존 방법들의 성능을 객관적으로 평가하고 비교할 수 있는 표준화된 기준을 제공하기 때문에 매우 중요하다고 할 수 있습니다.
이번 학회에서 Diffusion 모델과 더불어 GFlowNet 관련 후속 연구가 다수 있었다는 점 또한 주목할 만합니다.
GFlowNet은 분자를 다루는 데 매우 적합한 생성 모델입니다. 따라서 이번 포스팅에서는 딥러닝 연구자들에게 지속적인 관심을 받고 있는 Diffusion 모델과 GFlowNet에 관련한 논문을 살펴볼 것입니다. 그리고 이전에 소개한 Uni-Mol의 후속 버전에 대한 연구와 함께, 최근에 큰 주목을 받고 있는 연구 분야인 Protein Language Model에 관한 논문 중 한 편을 소개하도록 하겠습니다.
위의 연구 주제들 중 이번 포스팅에서 깊이 있게 알아볼 논문은 다음과 같습니다.
- SubGDiff: A Subgraph Diffusion Model to Improve Molecular Representation Learning
- Genetic-guided GFlowNets for Sample Efficient Molecular Optimization
- Uni-Mol2: Exploring Molecular Pretraining Model at Scale
- MutaPLM: Protein Language Modeling for Mutation Explanation and Engineering
NeurIPS 2024 논문 미리 보기 1 - Diffusion Model 기반의 표현형 학습 모델
첫번째로 소개드릴 논문은 SubGDiff입니다. SubGDiff는 Diffusion Model을 기반으로 한 컨포머 생성 및 화합물의 물성 예측 모델입니다. 기존에도 GraphMVP, GeoSSL 등과 같이 3D 컨포머의 학습을 통해 분자의 표현형을 학습하는 모델이 있었습니다. 그러나 SubGDiff의 메인 아이디어는 기존의 컨포머 생성 모델과 달리 분자의 하위 구조를 이용하는 데 있습니다.
기존의 연구인 Torsional Diffusion의 경우, 화합물의 결합 길이와 각도에 대한 정보를 사용하지 않았다는 한계가 있었습니다. 또한 같은 해에 발표된 GeoDiff의 경우, 각각의 원자들을 개별적으로 취급한다는 점에서 화합물의 특성을 제대로 반영하지 못했다는 한계가 존재했습니다.
SubGDiff의 연구진은 이러한 선행 연구들이 갖는 한계점에 주목하였습니다. 연구진은 화합물 내 존재하는 하위 구조 단위에 노이즈를 추가하는 아이디어를 적용했습니다. 아래 그림에서 보듯이, 노이즈를 추가하는 Forward 과정에서는 원자 단위로 좌표에 노이즈를 추가하는 것이 아니라, 하위 구조 단위로 노이즈를 추가하는 것을 볼 수 있습니다. 저자들은 이렇게 함으로써 Denoising 과정의 네트워크가 더 강한 표현력을 갖기를 기대했습니다.
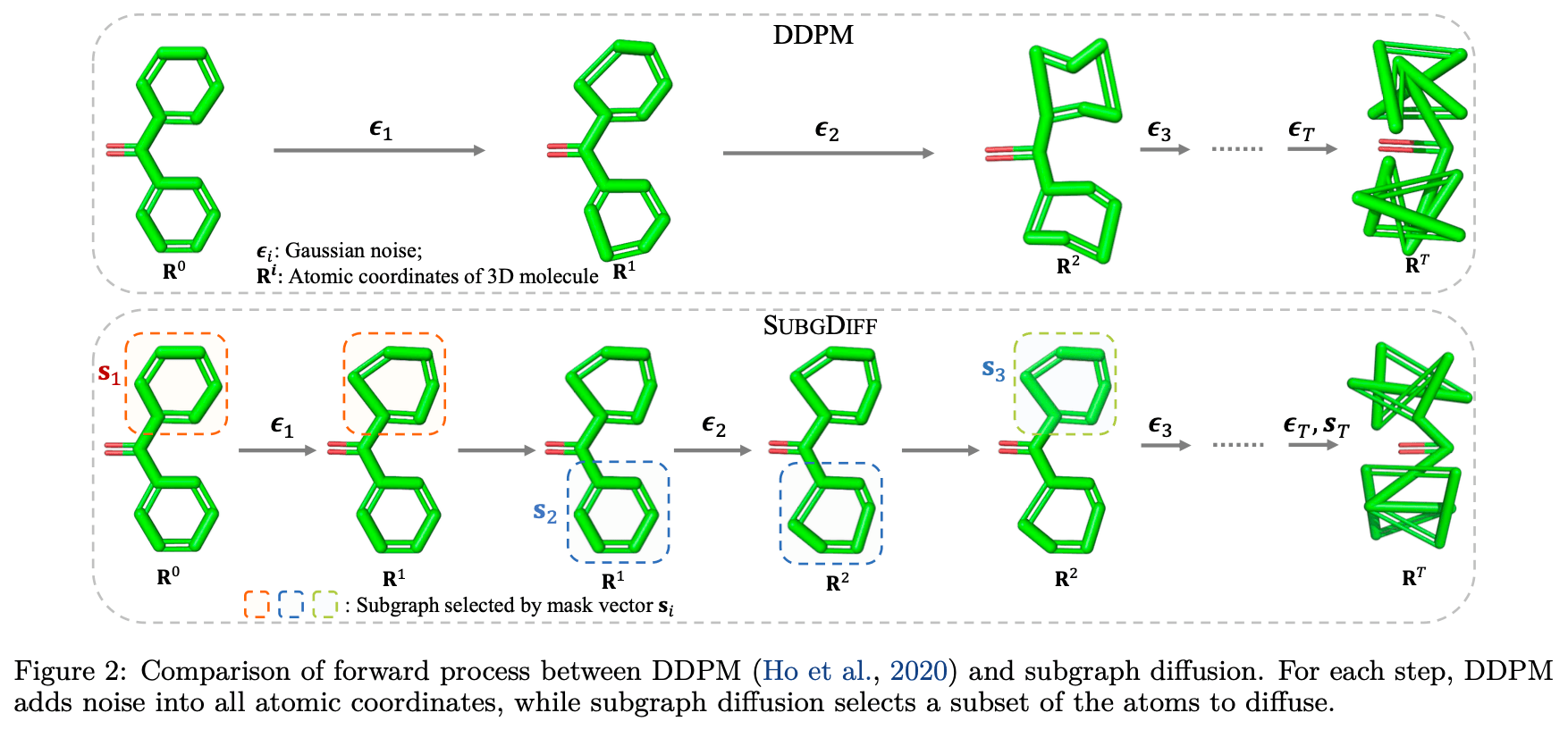
SubGDiff의 Forward 과정은 일반적인 Diffusion Model의 그것과 같습니다.
\[ q(R^t | R^{t-1}, s_t) = \mathcal{N} \left( R^t, \sqrt{1 - \beta_t \operatorname{diag}(s_t)} R^{t-1}, \beta_t \operatorname{diag}(s_t) I \right) \]
Forward 과정에서는 \(t-1\)단계에서 \(t\)단계로 갈 때 가우시안 노이즈를 추가하여 데이터를 점진적으로 변화시킵니다. 여기서 \(R_t\) 는 \(t\)단계에서의 3차원 좌표를 의미합니다.
다음은 Reverse 과정입니다. Reverse 과정에서는 Expectation State Diffusion 과정과 k-step same-subgraph diffusion 과정이 이용됩니다.
둘 중에 Expectation State Diffusion 과정에 대해서만 간단하게 살펴볼까요?
SubGDiff의 Forward 과정과 Denosing 과정을 수행하기 위해서는 다음과 같이 \(s_{1:t}\) 을 알아야 합니다.
\[ R^{t-1} = \mu_q(R^t, R^0, s_{1:t-1}, p_\theta(s_t | R^t)) \epsilon_\theta(R^t, t) + \sigma_q(t) z \]
여기서 \(\mu_q\) 는 다음과 같습니다.
\[ \mu_q(R^t, R^0, s_{1:t}) = \frac{1}{\sqrt{1 - \beta_t s_t}} \left( R^t - \frac{\beta_t s_t}{\sqrt{(1 - \beta_t s_t)(1 - \bar{\gamma}_{t-1}) + \beta_t s_t}} \right) \]
\[ \bar{\gamma}_t := \prod_{i=1}^t (1 - s_t \beta_t) \]
그러나 우리는 \(\mu_{q}\) 를 구하기 위해 필요한 \(s_{1:t}\)를 알 수 없습니다. 이것을 모른다면 각 Time Step에서 Denoising 과정을 정확하게 수행할 수 없습니다. 각 Time Step에서 축적된 Noise 의 분포를 제대로 학습하지 못했기 때문입니다.
따라서 \(\mu_{q}\)를 직접 구하는 대신에 \(\mu_{\hat{q}}\)으로 근사하여 Sampling 과정을 실시합니다. \(\mu_{q}\)를 \(\mu_{\hat{q}}\)로 근사하는 과정에서는 t 상태 직전까지의 상태, 그러니까 t가 0일 때부터 t-1일 때까지는 \(R^{t-1}\)의 기댓값을 사용하고, \(R^{t}\)일때는 위에서 살펴본 q를 그대로 사용하는 것이 핵심입니다.
\[ \mu_{\hat{q}}(R^t, R^0, s_{1:t}) := \frac{1}{\sqrt{1 - \beta_t s_t}} \left( R^t - \frac{s_t \beta_t}{\sqrt{\hat{s}_t \beta_t + (1 - s_t \beta_t)p^2 \sum_{i=1}^{t-1} \frac{\bar{\alpha}_{t-1}}{\bar{\alpha}_i} \beta_i}} \right) \]
\[ R^{t-1} = \mu_{\hat{q}}(R^t, R^0, s_{1:t-1}, p_\theta(s_t | R^t)) \epsilon_\theta(R^t, t) + \sigma_{\hat{q}}(t) z \]
이를 통해 각 Time Step에서의 Denoising 과정을 수행할 수 있게 됩니다.
SubGDiff는 위와 같은 방법으로 3D Conformation 정보를 이용하여 강한 표현력을 가지는 화합물의 표현형을 학습할 수 있었습니다. 이렇게 SubGDiff가 표현형을 학습하는 과정에서 습득한 컨포머 생성 성능은 다음과 같습니다. Diffusion Model을 사용한 다른 모델인 GeoDiff보다 월등히 좋은 성능을 보여줍니다.

이렇게 사전 학습으로 학습된 화합물의 representation을 이용해 화합물의 다양한 물성 예측도 가능합니다. SubGDiff는 BBBP, Tox21을 비롯하여 총 8개의 벤치마크 데이터세트에 대해 SOTA를 달성했습니다. SubGDiff는 소스 코드가 공개되어 여기서 확인할 수 있습니다.
NeurIPS 2024 논문 미리 보기 2 - 유전 알고리즘과 GFlowNet의 결합
세 번째로 소개드릴 논문은 Genetic GFN입니다. 앞서 설명드린 것처럼, GFlowNet을 활용한 논문입니다.
여기서 잠시 GFlowNet에 대해 알아보시죠. GFlowNet은 Flow Network based Generative Models for Non-Iterative Diverse Candidate Generation이라는 제목으로 2021년 NeurIPS에서 처음 소개된 연구이지만, 현재까지도 지속적인 관심을 받고 있습니다. GFlowNet은 생성 모델이며, 최근에는 Diffusion 모델과 함께 화합물 생성 분야에서 큰 두각을 드러내고 있습니다. 오늘은 GFlowNet에 대해 간단히 알아보고, 차후에 기회가 되면 히츠 블로그에서 다시 한 번 다루도록 하겠습니다.
간단히 설명하자면, GFlowNet은 화합물의 작용기를 단계적으로 붙여 가며 각 작용기가 붙을 확률을 학습하는 모델이라고 생각하면 됩니다.
아래 그림을 보시죠. 상태를 의미하는 각 노드에서 다음 노드로 나아감에 따라 화합물의 작용기가 하나씩 추가됩니다. 그리고 새롭게 추가된 노드가 선택될 확률을 Flow라 합니다. 이 Flow는 하나의 노드(상태)로 들어가는 Flow의 양과, 그 노드에서부터 나오는 Flow의 양이 항상 같아야 하기에 이런 이름이 붙었습니다. 그리고 가장 중요한 GFlowNet의 학습 목적은, 아래 트리의 루트에서 리프까지 특정 화합물이 생성될 확률이 그 화합물의 보상과 비례하도록 만드는 정책 함수를 학습하는 것입니다.
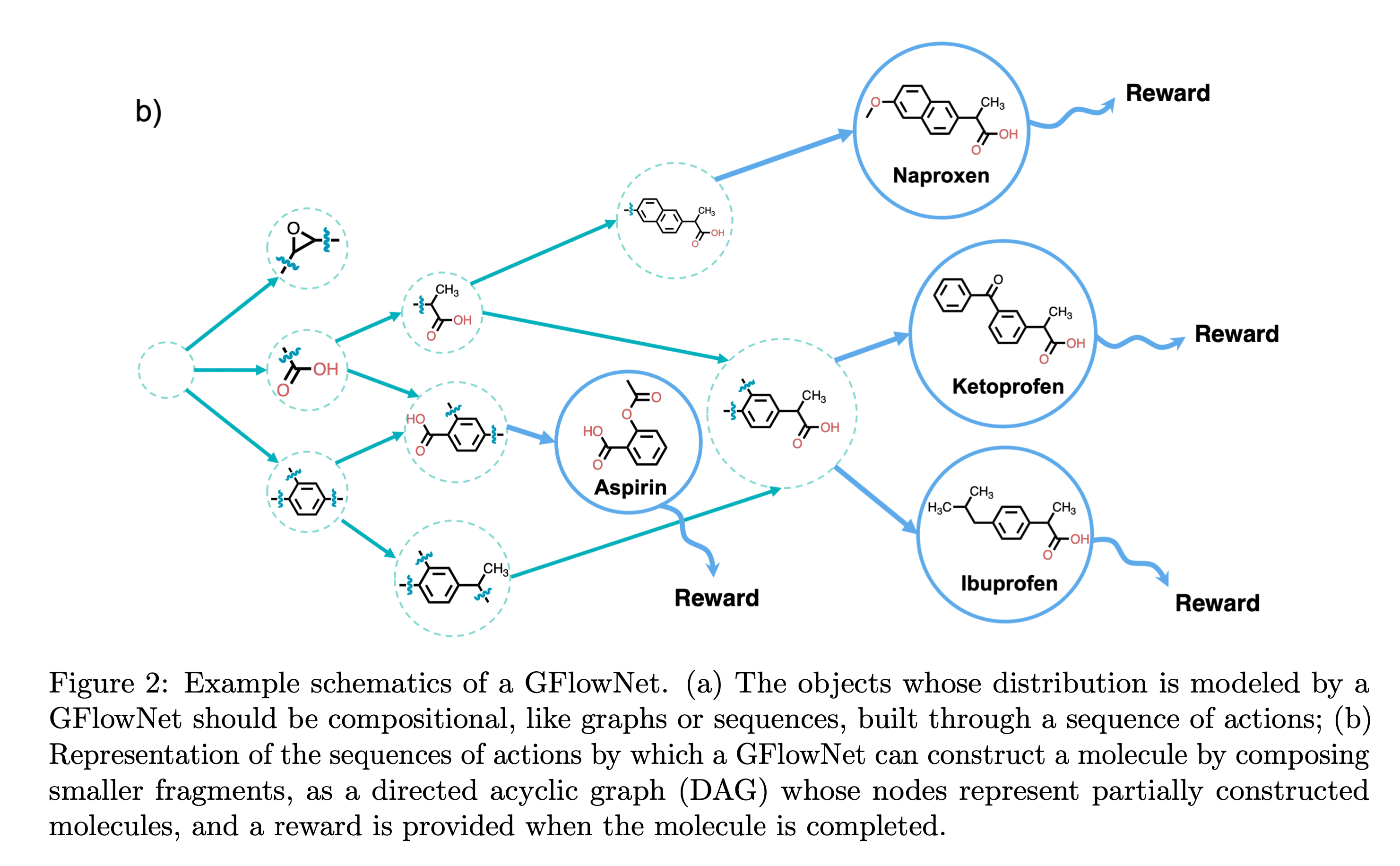
GFlowNet에서 정책 함수는 두 가지가 있습니다. 처음 상태인 아무것도 없는 상태에서 작용기를 하나씩 붙여 나아가며 목표 상태에 도달하는 정방향의 확률 분포 \(P_F\)와, 그 반대 과정인 역방향의 확률 분포 \(P_B\).이렇게 두 가지입니다. 이 두가지 정책 함수를 이용해서 다음의 식을 만족시키도록 정책 함수를 학습시키는 것입니다.
\[ Z \prod_{t=1}^n P_F(s_t \mid s_{t-1}) = R(s_T) \prod_{t=1}^n P_B(s_{t-1} \mid s_t) \]
Genetic GFN을 이해하기 위해서는 이 정도만 알면 됩니다.
자, 다시 Genetic GFN 설명입니다. Genetic GFN은 GFlowNet과 유전 알고리즘을 결합하여 화합물 디자인의 새로운 방식을 제안하였습니다. Genetic GFN의 구조는 다음과 같이 크게 두 부분으로 나눌 수 있습니다.
1. SMILES 생성
연구진은 위에서 설명한 GFlowNet의 예시와는 달리, 각 상태에서 작용기를 추가하는 방식이 아니라 문자열 기반인 SMILES를 이용했습니다. RNN 기반 모델로 현재 SMILES의 다음 토큰을 생성하여 다음 노드(상태)를 만들고, 이때 새로운 SMILES 토큰이 생성되는 확률을 정책 함수로 학습하도록 하였습니다. 이렇게 새로 생성된 SMILES는 다음 단계인 유전 알고리즘 단계에서 다양성을 늘리는 기반이 됩니다.
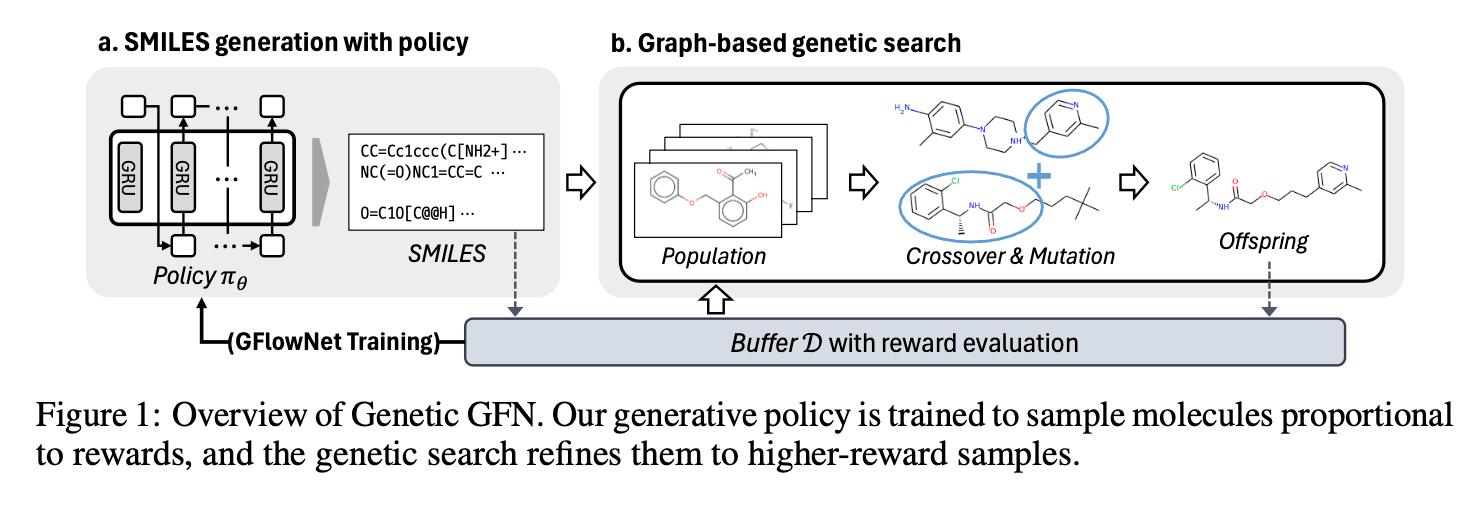
2. 유전 알고리즘 적용
연구진은 GFlowNet의 한계를 보완하기 위하여 유전 알고리즘을 사용하였습니다. 이는 화합물의 다양성을 높이고, 보상이 높은 지역을 효율적으로 탐색하기 위함입니다.
RNN으로 생성한 수많은 후보 화합물들을 가지고 다양한 Crossover, Mutation을 일으켜 새로운 화합물을 생성하도록 하였습니다. 연구에서 사용된 유전 알고리즘은 A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space라는 연구에 기반하였으며, 유전 알고리즘에 사용한 규칙은 아래 그림과 같습니다.
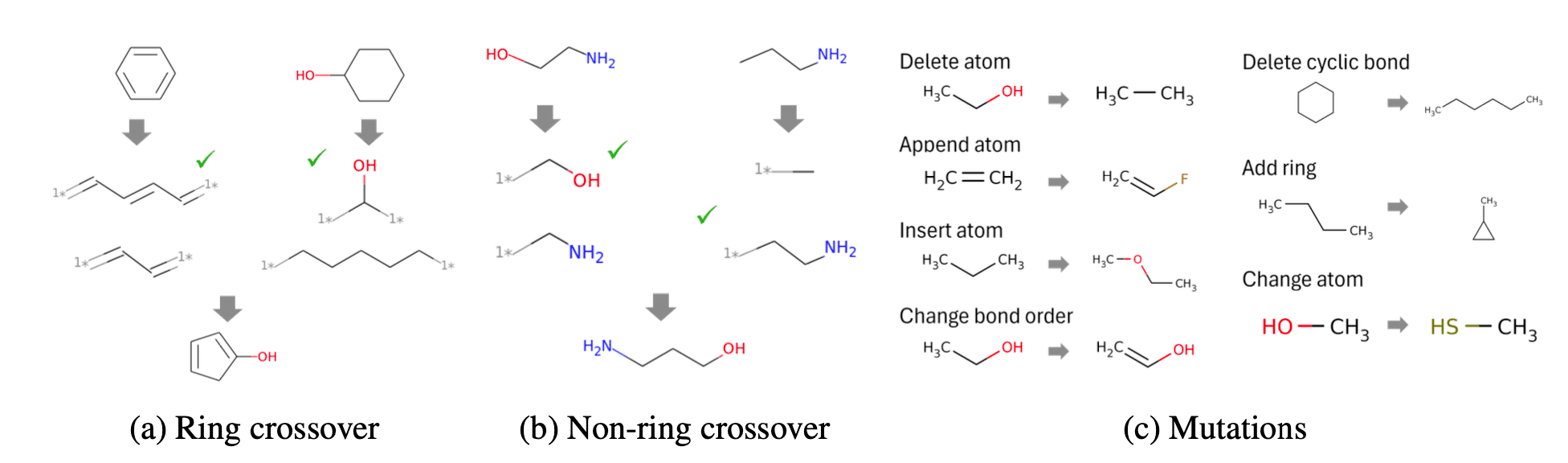
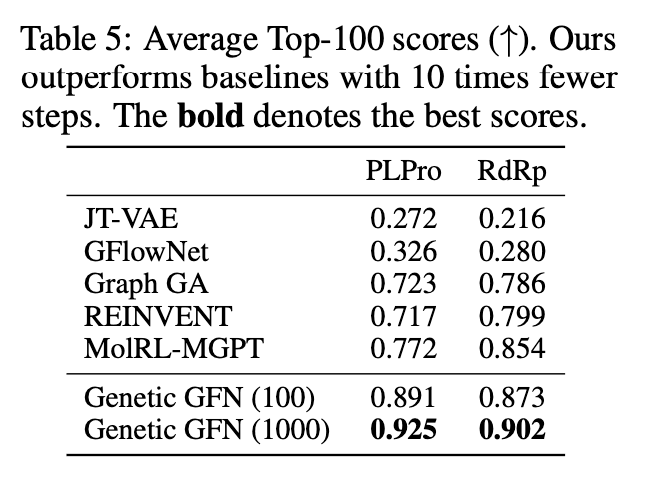
연구진은 Genetic GFN의 성능을 평가하기 위해 SARS-CoV-2 억제제 설계 실험을 진행하였습니다. 그 결과, 두 가지 타겟 단백질에 대한 성능이 기존 연구들의 성능 지표보다 높다는 것을 확인할 수 있었습니다.
NeurIPS 2024 논문 미리 보기 3 - Transformer 기반의 표현형 학습 모델
두번째 논문은 Uni-Mol2에 대한 논문입니다. Uni-Mol은 얼마전 히츠의 블로그에도 소개된 적이 있는 모델입니다. (링크)
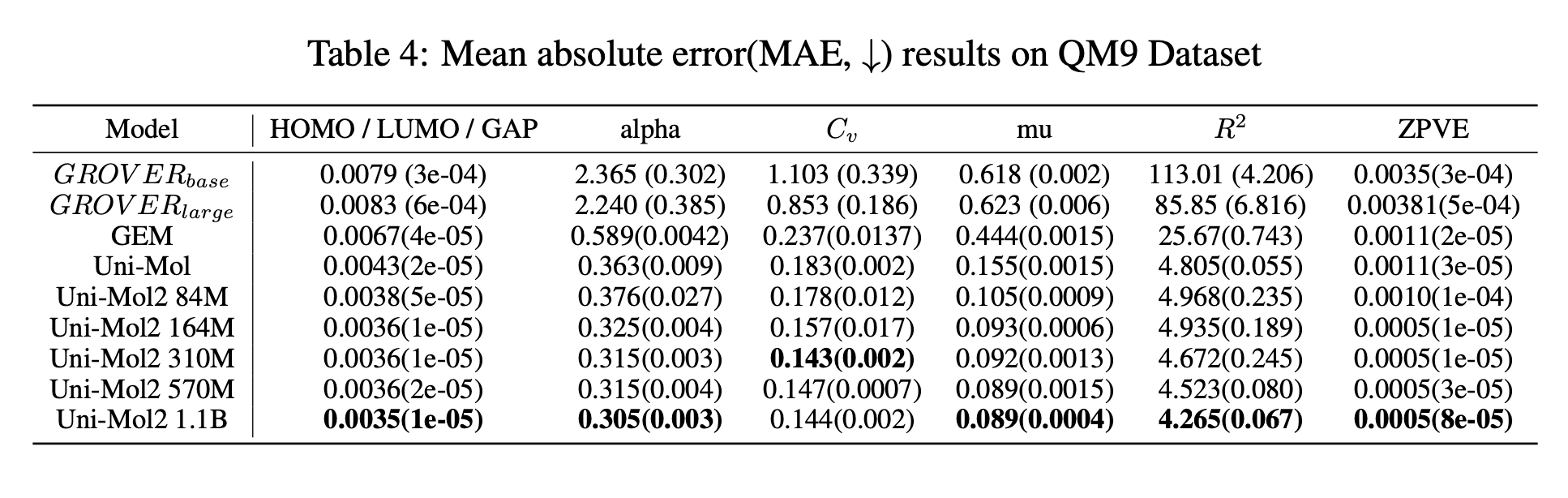
그 Uni-Mol이 Uni-Mol2로 개선되어 2024 NeurIPS에 소개되었습니다. Uni-Mol2는 8억 개의 화합물 구조를 11억 개의 파라미터를 이용해 예측하도록 사전 학습되었습니다. 기존 Uni-Mol에서 사용한 1,900만 개의 화합물 구조에 비하면 40배 이상의 학습 데이터를 사용한 것입니다. 학습 데이터는 기존 Uni-Mol에서 사용한 데이터 대부분과 ZINC 20 데이터세트를 활용하여, 학습 데이터의 규모를 비약적으로 늘렸습니다.
Uni-Mol2의 아키텍처 자체는 기존 Uni-Mol의 아키텍처에서 크게 달라진 점은 없지만, 다음과 같은 주요 기여를 이루었다고 설명합니다.
- 거대한 데이터세트 이용: 연구진은 약 8억 8천 4백만 개의 3D 분자 구조를 포함한 데이터세트를 준비했으며, 이들 분자 구조는 7천 3백만 개의 다양한 골격(scaffold)을 포함하고 있습니다. 이는 현재까지 가장 큰 3D 분자 구조 데이터세트로, 대규모 분자 모델을 학습하는 데 필수적인 기반 데이터로 활용될 여지가 있다고 합니다.
- Scaling law 입증: Uni-Mol2의 확장성과 유연성을 평가하기 위해, 모델의 파라미터 수(8천 4백만 개에서 11억 개), 데이터세트 크기, 컴퓨팅 리소스에 따른 검증 손실(validation loss) 간의 관계를 체계적으로 분석했습니다. 이는 분자 사전 학습에 대해 확장 법칙(scaling law)을 입증한 첫 사례라고 하며, Uni-Mol2는 지금까지 발표된 분자 사전 학습 모델 중 최대 규모로 주목받고 있습니다.
- 다운스트림 작업에 대한 성능 분석: Uni-Mol2는 모델 파라미터 수가 증가할수록 다운스트림 작업 성능이 꾸준히 향상됨을 보여줍니다. 특히 11억 개의 파라미터를 가진 모델은 기존 방법에 비해 상당한 성능 개선을 달성했습니다.
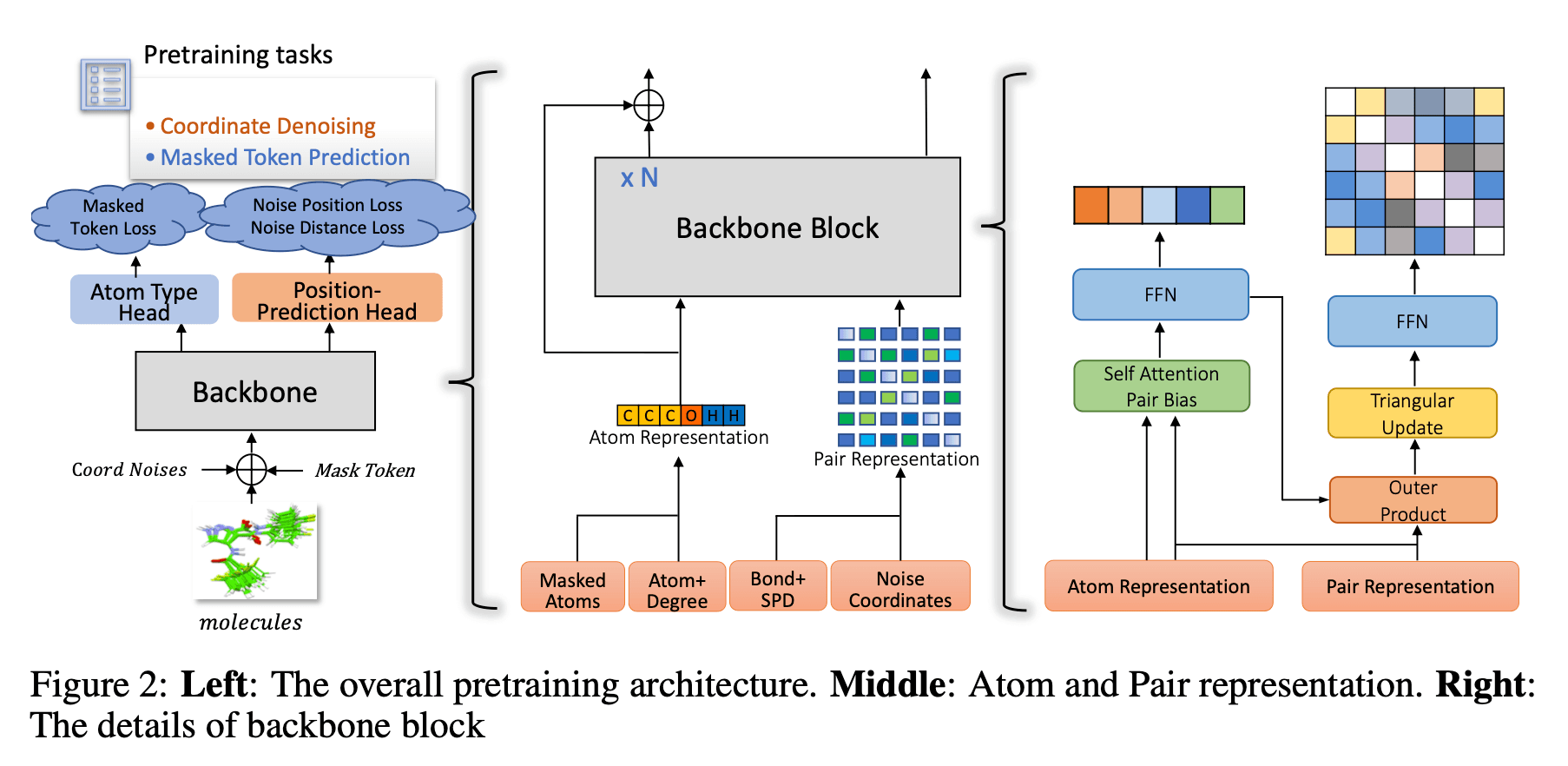
Uni-Mol2의 사전학습 과정은 다음과 같습니다.
-
1. Uni-Mol2의 학습에는 약 8억 개의 학습 데이터가 사용되었으며, 각 데이터의 컨포머는 RDKit의 ETKDG로 생성되었고, MMFF 방식으로 최적화되었습니다.
-
2. 원자의 좌표에 노이즈를 추가하는 과정에서는 가우시안 분포를 따르는, 표준편차가 0.2 Å인 좌표값을 임의로 더합니다.
-
원자 토큰에 대해서는 무작위로 15%를 마스킹 처리합니다.
-
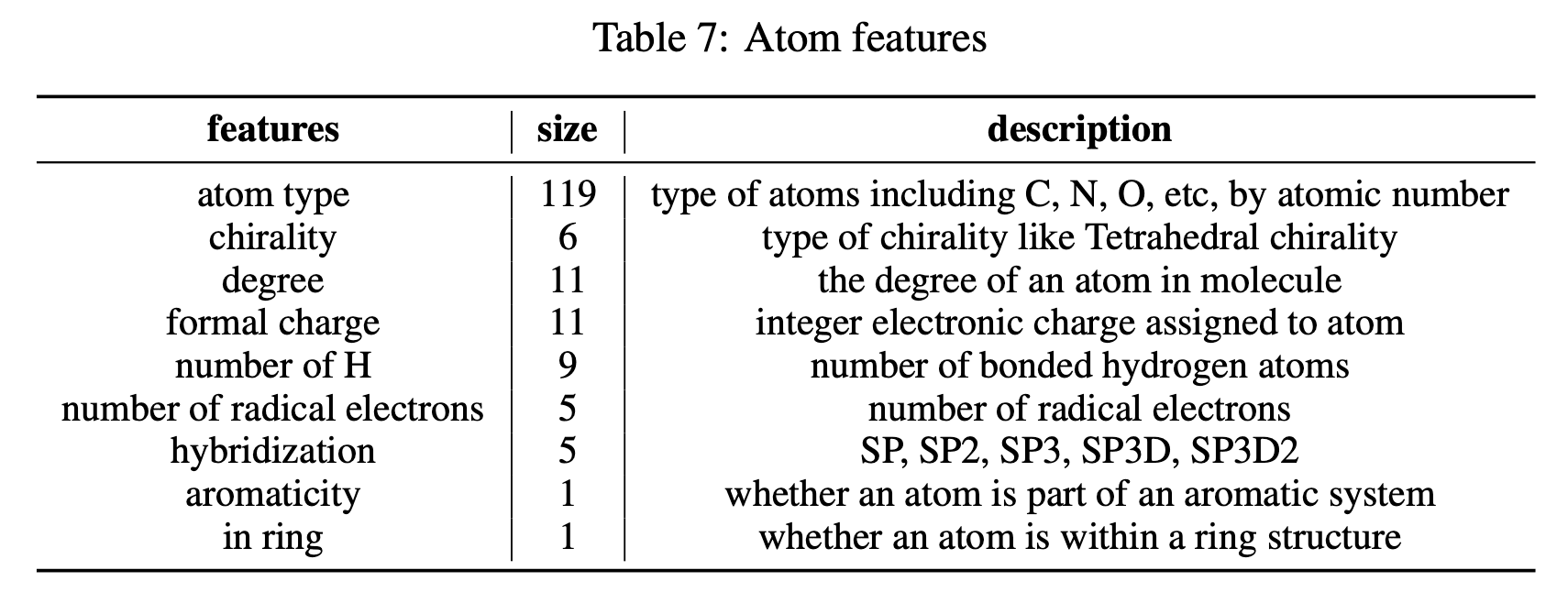
마지막으로, 원자 특징(Table 7), 결합 특징(Table 8), 최단거리(SPD; Floyd–Warshall 알고리즘으로 계산) 특징을 50% 확률로 마스킹합니다.
-
- 3. Backbone 블록은 N개로 구성되며, 각 블록에서는 원자의 표현형과 pair의 표현형을 학습합니다. 각 블록의 구성은 위 그림과 같습니다.

간단히 요약하면, Uni-Mol2는 학습 데이터인 화합물의 좌표값과 원자 종류, 결합 거리 등을 마스킹하여 진행되는 자기지도 학습입니다. 그렇다면 당연히 그 과정에서 SubGDiff와 마찬가지로 화합물의 표현형을 학습하겠죠? 맞습니다. 그래서 Uni-Mol2에서도 화합물의 물성 예측을 통해 화합물의 표현형 학습 성능을 확인했습니다. 다음 결과처럼, Uni-Mol2는 연구에서 제시된 모든 벤치마크에서 우수한 성능을 보였습니다.
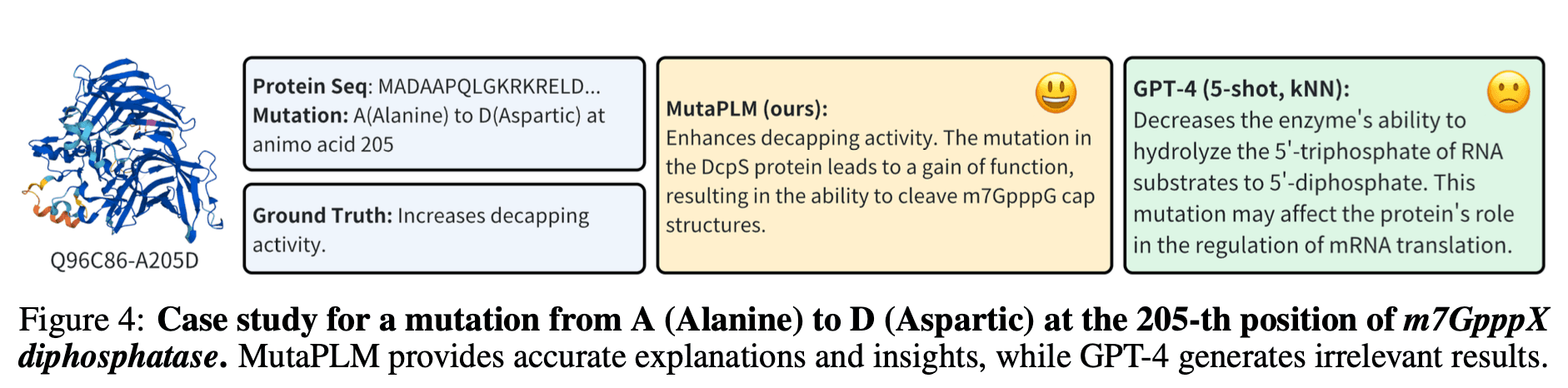
NeurIPS 2024 논문 미리 보기 4 - 단백질 언어 모델을 이용해 단백질의 변이를 자연어로 설명하기
단백질 언어 모델(Protein Language Model, PLM)은 단백질의 1차 서열을 자연어처럼 다루어 학습하는 모델입니다. 언어 모델의 개념을 단백질 서열 분석에 적용한 것으로, 자연어의 단어와 같이 아미노산 서열을 마치 “단어”처럼 인식하여 단백질의 구조나 기능을 예측하고 분석하는 데 사용됩니다. 단백질 언어 모델의 주요 아이디어는, 단백질의 아미노산 서열을 이해하면 단백질의 구조적·기능적 정보를 효과적으로 예측할 수 있다는 것입니다.
논문에서 제시한 MutaPLM은 단백질의 1차 서열에서 아미노산이 돌연변이로 바뀌었을 때, 그 변화가 단백질의 활성 및 기능에 미치는 영향을 자연어로 설명해 주는 모델입니다.
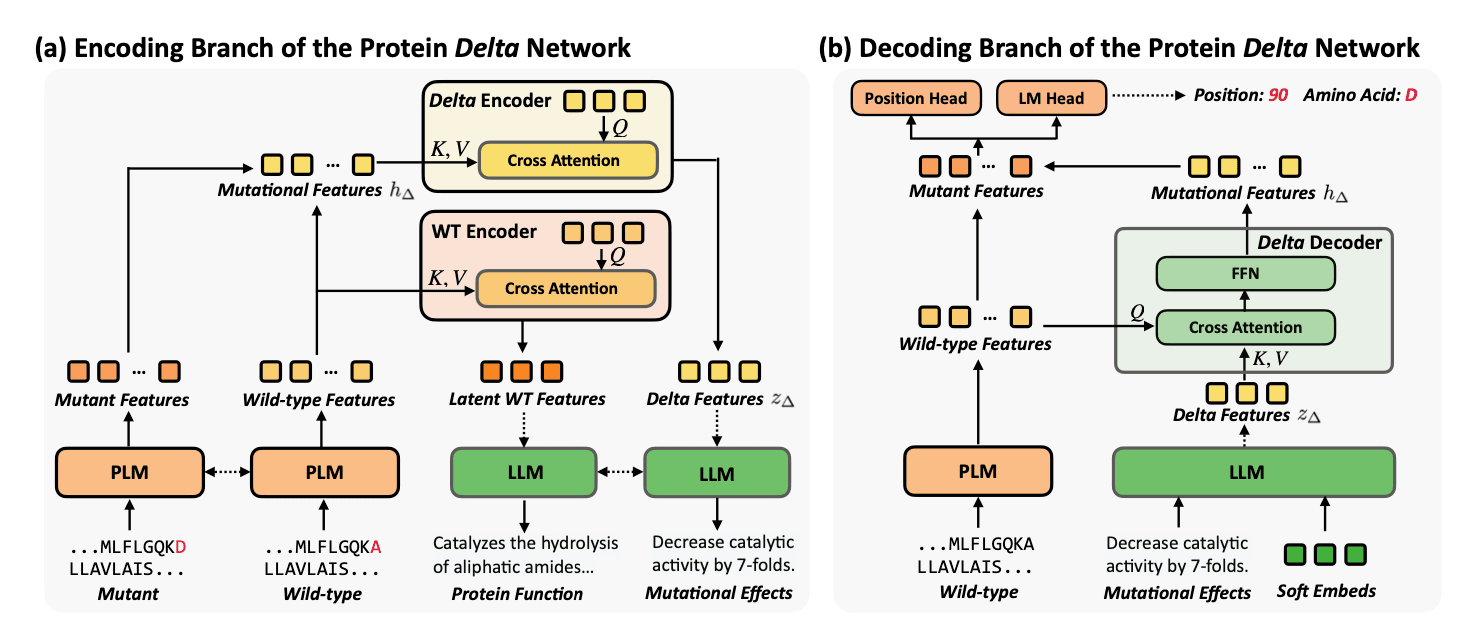
MutaPLM은 '단백질 델타 네트워크'라는 이름의 네트워크를 이용합니다. 이 네트워크는 아래 그림과 같은 구조로 설계되어 있습니다. 보시는 것처럼, 이는 인코더-디코더 구조입니다.
먼저 인코더에서는 단백질의 원형(Wild-type)과 변이형(Mutant) 아미노산 서열을 입력으로 받습니다. 입력으로 받은 각각의 아미노산 서열을 PLM의 입력으로 넣어 각각의 표현형을 얻고, 이를 각각의 인코더에 입력하면 돌연변이에 대한 많은 정보를 포함하고 있는 표현형을 얻을 수 있습니다. 이 표현형은 LLM에 전달되어, 변이가 가지는 영향을 자연어로 생성합니다. 여기서 사용되는 LLM은 기존에 출간된 과학 문헌들을 통해 학습된 모델로, 연구진이 직접 학습하였다고 합니다.
다음은 디코딩 과정에 대한 설명입니다. 디코딩 과정에서는 원형 단백질 서열과 LLM이 생성한 변이에 대한 설명을 입력으로 사용하여 인코더 단계의 입력을 복원합니다. 그리고 인코더의 입력과 디코더의 출력 간 차이를 Weighted Cross-Entropy 손실 함수를 통해 계산하여 학습됩니다.
연구진은 MutaPLM에 사용되는 언어 모델을 학습시키기 위해 MutaDescribe라는 데이터세트를 직접 구축했습니다. 이 MutaDescribe 데이터세트는 MutaPLM을 파인튜닝(Fine-tuning)하기 위해 사용됩니다. 데이터세트에는 약 2만여 개의 단백질과 그들의 점돌연변이 서열이 포함되어 있으며, 해당 돌연변이의 효과가 자연어로 기술되어 있습니다. MutaDescribe 데이터세트는 허깅페이스(Hugging Face)에 업로드되어 있어 직접 확인하실 수 있습니다.
연구진은 ESM-2로 알려진 기존의 Protein Language Model에 비해 ROUGE-L, BLEU-2 벤치마크에서 성능의 우위를 보였으며, 이는 SOTA 수준의 성능이라고 주장하였습니다.
NeurIPS 2024에서의 신약 개발과 AI의 미래
NeurIPS 2024는 신경망과 인공지능 기술이 과학과 산업에 실질적으로 기여하는 다양한 사례를 보여주는 중요한 장이 되고 있습니다. 특히 신약 개발과 분자 설계 분야에서 AI의 활용 가능성을 엿볼 수 있는 다양한 연구들이 발표되면서, AI 기술이 의료와 생명과학의 혁신을 주도하고 있음을 확인할 수 있었습니다.
이번 포스팅에서 다룬 Diffusion 모델 기반의 SubGDiff, 유전 알고리즘과 결합된 GFlowNet, 확장된 Uni-Mol2, 그리고 단백질 언어 모델인 MutaPLM은 AI 연구가 신약 개발 분야에 얼마나 심도 깊고 넓은 영향을 미치고 있는지를 보여줍니다. 이러한 연구들은 화합물 설계의 효율성을 높이고, 단백질 구조와 변이를 더욱 정밀하게 이해하도록 도와줌으로써 새로운 치료제 개발의 속도와 가능성을 크게 향상시키고 있습니다.
AI 기술은 이제 단순히 이론적 연구를 넘어, 실제로 복잡한 문제를 해결하는 데 사용되는 실질적인 도구로 자리 잡았습니다. 특히 NeurIPS와 같은 학회에서 다뤄지는 연구들은 AI 기술의 경계를 확장하고, 기존 과학적 방법론을 보완하며, 미래의 새로운 가능성을 열고 있습니다. 앞으로도 NeurIPS와 같은 플랫폼에서 AI와 신약 개발 분야가 어떻게 협력하며 발전해 나가는지를 주목할 필요가 있습니다. AI와 과학의 융합이 만들어낼 놀라운 성과들을 기대하며, 더 나은 미래를 위한 새로운 아이디어와 연구들이 계속해서 나오길 바랍니다.