30년 전 개발된 AlphaFold의 비밀 무기


You’re more powerful than you think
한 사람이 자신의 진정한 가치를 인정받기까지 시간이 걸리곤 합니다. 인류 역사상 가장 혁명적인 기업가라고 불리는 스티브 잡스 역시 자신이 공동 창립한 애플에서 쫓겨나는 시련을 겪었습니다. 그는 애플이 위기에 처한 후 다시 복귀하여 iMac, iPod, iPhone, iPad 등의 혁신적인 상품을 연달아 성공시키며 결국 본인의 가치를 증명할 수 있었습니다. 잡스의 인생처럼, 어떤 사람의 성공 스토리 뒤에 파란만장한 삶이 숨어 있는 것은 그리 놀라운 일이 아닐 것입니다.

2024년 노벨 화학상 수상의 영예를 안은 AlphaFold에게도 이런 파란만장한 뒷이야기가 숨겨져 있습니다. 단백질의 3차원 구조를 예측해주는 혁신 딥러닝 기술인 AlphaFold는 다중 서열 정렬(Multiple Sequence Alignment; MSA)이라는 기술을 사용해 정확도를 비약적으로 향상시켰습니다. MSA는 보통 사람들에게는 친숙하지 않은 이름이지만, 사실 생물정보학(Bioinformatics) 커뮤니티에서는 단백질의 기능 예측을 위해 사용되던 전통적인 기술입니다. MSA의 조상격인 ClustalW가 1994년에 개발되었으니, MSA의 근간은 30년 전인 1994년에 이미 완성되었다고 보아도 과언이 아닙니다.
AlphaFold의 성공으로 인한 스포트라이트가 비춰지기 전에 MSA는 AlphaFold 이전까지는 대중에게 알려지지 않았음은 물론, 생물정보학 커뮤니티에서도 서서히 주목도가 떨어져 가던 기술이었습니다. 이런 MSA가 다시금 중요하게 떠오른 이유는 무엇일까요? 이번 포스팅에서는 MSA가 무엇인지 과거와 현재를 돌아보고, 어째서 30년 전 기술이 AlphaFold의 성능을 비약적으로 끌어올리는 데 큰 역할을 할 수 있었는지 이야기해보겠습니다.
Multiple Sequence Alignment (MSA)란 무엇인가?
MSA는 셋 이상의 생물학적 서열 (DNA, RNA, 단백질 등)을 정렬하여 공통 조상, 기능적 영역, 그리고 진화적 관계를 분석하는 방법입니다. 이번 포스팅에서는 단백질을 중심으로 말씀드리겠습니다. 이렇게 말하면 어렵게 느껴질 수도 있겠지만, 쉽게 말해 여러 단백질 서열들에서 공통적으로 나타나는 부분을 찾아내는 과정입니다. 즉, MSA는 여러 단백질의 아미노산 서열을 한 번에 정렬함으로써 각 서열 간의 유사성과 차이를 한눈에 파악할 수 있는 기술입니다.

생명 현상을 매개하는 중개자 역할을 한다고 할 수 있는 단백질은 수십 개에서 수천 개의 아미노산 서열로 이루어집니다. 이 아미노산 서열은 생명체의 진화 과정에서 유전자의 돌연변이로 인해 변경될 수 있습니다. 그렇다면, 만약 여러 종에 걸쳐 서열이 보존된 부분이 있다면 어떨까요?
이러한 보존된 영역은 단백질의 주요 기능과 연관이 있을 확률이 높습니다. 반면, 서열이 보존되지 않은 영역은 생명 현상을 유지하는 데 큰 영향이 없을 확률이 높습니다. 만약 생명 현상 유지에 필수적인 서열이 변경되어 제 기능을 하지 못하게 되었다면, 그 종은 자연 선택으로 인해 도태되었을 것이기 때문입니다. 특정 유전자나 단백질 서열이 여러 종에서 일관되게 보존되어 있다면, 이는 해당 서열이 중요한 기능적 역할을 할 가능성이 높다는 것을 시사합니다. MSA로부터 얻은 결과는 단백질 기능 연구 외에도 질병의 유전적 원인 분석, 진화계통학적 분석 등 다양한 생물학적 연구에 활용될 수 있습니다.
MSA의 흥망성쇠(興亡盛衰)
MSA의 역사에 대해서 일일이 나열하려면 포스팅 한 개로는 부족합니다. 그러니 이번에는 MSA의 발전에 중요한 역할을 한 몇 가지 기술들만 간단히 언급하도록 하겠습니다. MSA의 개념 자체는 기존에도 있었지만, 중요한 전환점을 맞는 첫 신호탄은 1985년에 쏘아올려졌습니다.
1985년: FASTA 형식의 등장
FASTA는 빠르다는 뜻의 “Fast”와 Alignment의 앞글자를 따서 만들어졌습니다. FASTA는 원래 서열 정렬 프로그램이었기 때문에 서열 정렬을 빠르게 수행할 수 있다는 점을 강조하고자 이러한 이름이 붙여졌습니다. 이때 FASTA의 입력으로 사용된 파일 포맷이 오늘날에도 FASTA 형식(.fasta)으로 널리 사용되고 있습니다. FASTA 형식은 간단하면서도 효율적인 구조로 설계되었으며, 아래 두 가지 요소로 이루어져 있습니다.
- 헤더 라인:
>로 시작하며 서열의 이름이나 설명이 포함. - 서열 데이터: 연속적으로 작성된 단백질(또는 핵산) 서열.
👨🏻💻UniProt에서 다운로드 받은 P0DK86의 fasta 파일 형식
P0DKB6 RecName: Full=Mitochondrial pyruvate carrier 1-like protein; MARMAVLWRKMRDNFQSKEFREYVSSTHFWGPAFSWGLPLAAFKDMKASPEIISGRMTTA LILYSAIFMRFAYRVQPRNLLLMACHCTNVMAQSVQASRYLLYYYGGGGAEAKARDPPAT AAAATSPGSQPPKQAS
빠르게 많은 서열들을 정렬하는 것이야말로 MSA의 가장 핵심적인 요소이기 때문에, FASTA 형식을 MSA의 첫 번째 열쇠로 봐도 무리가 아닙니다.
1990년: BLAST의 개발
BLAST(Basic Local Alignment Search Tool)는 지역 서열 정렬(Local Alignment)을 통해 주어진 서열과 데이터베이스 내 다른 서열 간의 유사성을 찾아내는 서열 정렬 도구입니다. BLAST는 전체 서열이 아닌 서열의 가장 유사한 부분만을 정렬하기 때문에 기존의 정렬 알고리즘보다 훨씬 빨랐습니다. BLAST 알고리즘은 짧은 word 단위를 기반으로 시드를 탐지하고, 시드 확장을 통해 지역적으로 높은 유사성을 가진 영역을 찾음으로써 전체 서열보다는 중요한 부분에 더 초점을 맞출 수 있었습니다.
BLAST는 기본적으로 두 서열 사이의 유사도를 판별하는 Pairwise Sequence Alignment(PSA) 도구이지만, 추후 개발되는 MSA 알고리즘을 보조하는 데 중요한 역할을 합니다. 크게 두 가지 정도로 요약할 수 있겠습니다.
- MSA를 수행하기 전에 MSA 대상 서열과 비슷한 서열들을 거대 단백질 데이터베이스에서 빠르게 검색하여 추출합니다.
-
BLAST 결과를 기반으로 서열을 클러스터링하여, 서열들의 진화적 연관성을 바탕으로 MSA 알고리즘이 효율적으로 작동하도록 돕습니다.
1994년: ClustalW, MSA의 표준을 제시하다
ClustalW는 생물정보학에서 매우 널리 사용된 프로그램입니다. ClustalW의 “W”는 Weighted라는 의미인데, 이는 ClustalW가 서열 간의 유사성을 기반으로 한 가중치를 부여하는 방식으로 작동하기 때문입니다. ClustalW는 점진적 정렬(Progressive Alignment)이라는 방법을 사용했는데요. 처음에는 Pairwise 정렬을 수행하고, 이를 기반으로 점진적으로 Multiple 정렬을 생성하는 방식입니다. 뿐만 아니라 서열 사이의 유사성에 따라 진화적 관계를 나타내는 가이드 트리(Guide Tree)를 생성하고, 이 가이드 트리를 기반으로 정렬 순서를 결정했습니다. 이러한 개념들은 이후 개발된 MSA 프로그램들(e.g. Clustal Omega, MUSCLE, MAFFT 등)에 큰 영향을 미쳤으며, 오늘날의 MSA 도구에서도 널리 사용되는 원리입니다.
비록 현재는 더 빠르고 정교한 도구들이 널리 사용되고 있지만, ClustalW는 이렇듯 후속 알고리즘에 미친 영향이 커 MSA의 표준을 제시한 역사적 이정표로 여겨지고 있습니다. 제목에서 AlphaFold의 비밀 무기가 30년 전에 개발되었다고 표현한 것이 바로 이 ClustalW를 기준으로 하고 있습니다. 심지어 ClustalW는 사용자 인터페이스도 우수했습니다. 이를 통해 MSA 기반 연구는 황금기를 맞이하였으며, MSA는 단백질의 기능을 연구하는 데 필수적인 도구로 자리매김했습니다.
NGS와 딥러닝이 불러온 MSA의 위기
잘 나가던 MSA 연구는 2000년대와 2010년대에 크나큰 도전에 직면하게 됩니다.
2000년대에는 Next-Generation Sequencing(NGS) 기술의 발전으로 인해 엄청난 양의 서열 데이터가 쏟아지기 시작했습니다. 기존의 MSA 도구는 서열 수가 많아질수록 계산에 필요한 자원이 기하급수적으로 증가했기 때문에, 대규모 서열 데이터를 처리하기에는 속도와 메모리 효율성이 부족하다는 한계를 드러냈습니다. 물론 단백질 도메인 분석, 보존된 유전자 영역 탐구 등 생물정보학적 정밀 분석에서는 MSA가 여전히 사용되었지만, 기술의 패러다임이 “빅데이터”로 이동하면서 대규모 서열 데이터 분석에서 MSA를 대체하거나 보완하기 위한 새로운 기술들이 등장하게 됩니다. 프로파일 기반 정렬, Hidden Markov Model(HMM), NGS 리드 매핑 기술, 데이터 클러스터링 기반 기술 등이 많은 주목을 받았습니다.
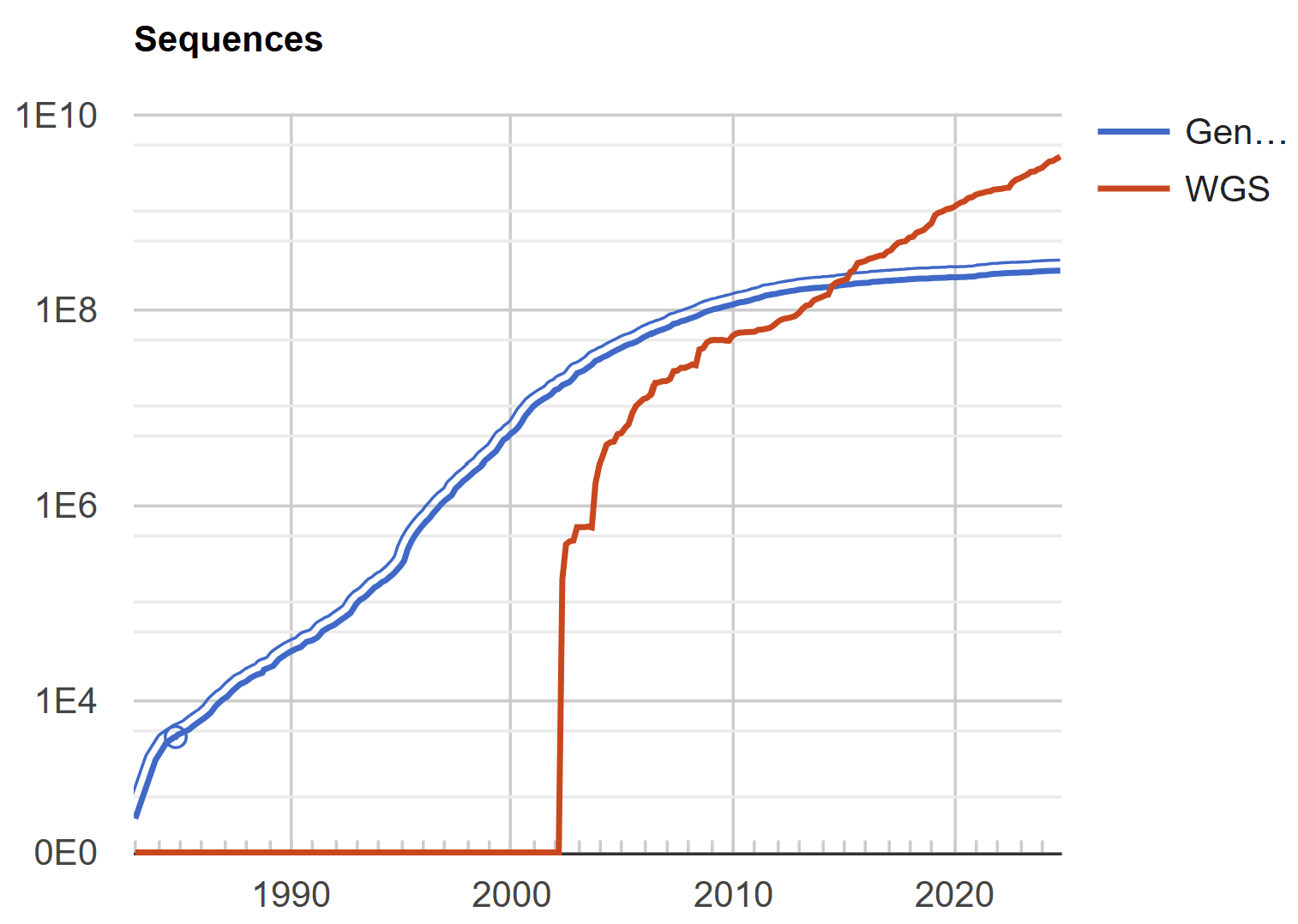
2010년대에는 딥러닝이 생물학적 서열 데이터를 분석하는 새로운 기술로 부상하면서 MSA는 또 다른 도전을 맞게 됩니다. MSA가 NGS의 등장으로 위기를 맞았던 것과는 반대로, 딥러닝은 엄청난 양의 데이터를 생산하는 NGS와 찰떡궁합이었습니다. 딥러닝 모델은 많은 데이터를 학습할수록 높은 예측 성능은 물론 일반화 능력까지 보여줬기 때문입니다. 딥러닝 모델들은 NGS에 의해 생성된 방대한 서열 데이터를 학습하면서, 기존 MSA보다 훨씬 더 효율적으로 서열 간의 패턴을 파악하고 예측할 수 있었습니다. 이들 모델은 대규모 데이터 학습을 통해 복잡한 생물학적 패턴을 발견하는 데 매우 효과적이었고, 계산 자원 측면에서도 MSA에 비해 더 나은 효율성을 보여주었습니다.
Recurrent Neural Networks(RNN)와 Convolutional Neural Networks(CNN) 같은 모델들은 서열 데이터를 이해하고 예측하는 데 적합했습니다. 이러한 딥러닝 기반 서열 특성 예측 모델의 성장은 2010년대 후반, 서열 데이터에 Transformer 기반 모델이 도입됨으로써 마침내 절정에 달했습니다. Transformer 기반 모델들은 MSA는 물론 RNN과 CNN 기반 모델들까지도 압도하는 성능을 보여주었습니다.
AlphaFold(알파폴드)와 함께 화려하게 부활한 MSA
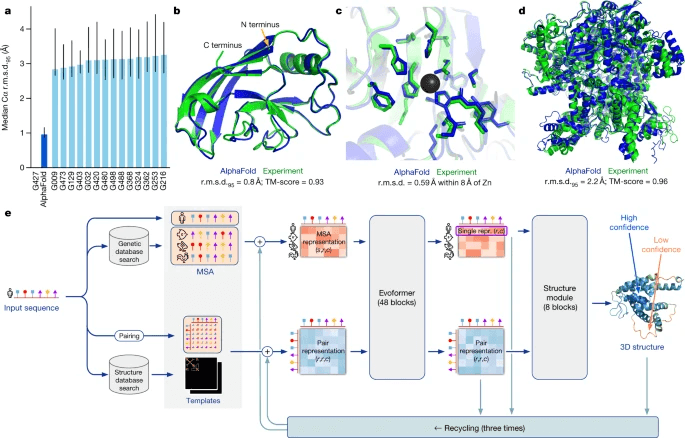
내가 무릎을 꿇었던 건 더 멀리 나아가기 위한 추진력을 얻기 위해서였다는 말이 있죠? 흥미롭게도 MSA를 위기로 몰고 갔던 딥러닝에 의해 MSA는 더욱 화려하게 부활하게 되었습니다. 딥러닝 기반 단백질 구조 예측 모델의 선두 주자인 AlphaFold는 MMSeqs2라는 MSA 모델을 통해 단백질 서열의 진화적 정보를 추출해 딥러닝 모델의 입력으로 사용합니다. MSA는 학습에서 크게 다음 두 가지 역할을 담당합니다.
-
진화적 정보 제공
MSA는 단백질의 구조적, 기능적 중요 부위를 식별할 수 있습니다. 진화적으로 보존된 영역은 단백질이 안정적인 3차원 구조를 유지하는 데 중요한 역할을 합니다. 알파폴드는 MSA를 통해 이러한 보존된 패턴을 학습합니다.
-
공진화적 (Co-evolutionary) 신호 추출
MSA를 통해 얻은 여러 서열 사이의 공진화적 (Co-evolutionary) 상호작용은 단백질 내 아미노산 간 접촉을 예측하는 데 사용됩니다. 가령 두 아미노산 잔기가 진화적으로 상호 의존적인 변이를 보인다면, 이들이 3차원 공간상에서 가까이 위치할 가능성이 높습니다. 알파폴드는 이런 신호를 학습하여 단백질의 접힘 구조를 모델링합니다.
단백질 서열들 간의 보존된 영역을 파악하고, 이를 통해 단백질의 기능적 특성을 이해하는 데 도움을 주는 MSA를 입력받음으로써 AlphaFold는 기존의 단백질 구조 예측 모델들과 비교할 수 없을 정도로 높은 성능을 보였습니다. 이를 바탕으로 아직 구조가 밝혀지지 않은 수만 개의 단백질 구조를 예측했고, 그 가치를 인정받아 2024년 노벨 화학상을 수상하게 됐습니다. AlphaFold의 성공은 MSA와 같은 전통적인 생물정보학 기법이 최신 딥러닝 기술과 결합될 때 얼마나 강력한 시너지를 일으킬 수 있는지를 보여줍니다. AlphaFold에 대한 보다 더 기술적인 이야기는 이 블로그 글에서 읽어보세요.
스포트라이트가 올 때까지
어떤 혁신적인 기술도 태동할 때에는 잘 알려져 있지 않다가, 나중에 그 진정한 가치가 발굴되며 큰 파란을 일으키는 경우가 많습니다. 유전학계에 일대 혁신을 일으킨 CRISPR-Cas9 기반 유전자 편집 기술도 1987년 처음 발견되었을 때에는, 그저 박테리아가 바이러스에 대항하기 위해 사용하는 흥미로운 방어 메커니즘에 불과했습니다.
오늘날 기술의 패러다임을 쥐고 있는 AI 역시 GPU를 비롯한 컴퓨터 하드웨어의 발전과 역전파 알고리즘의 발견 이전에는 마이너한 학문에 불과했습니다. 그러나 결국 이 기술들은 진정한 가치가 발굴되었습니다. MSA 역시도 30년 전 사람들이 생각했던 것보다 그 내재 가치가 컸기 때문에, 오늘날 새롭게 그 가치를 인정받은 것 같습니다.
히츠에서도 더 우수한 기술 개발에 매진하여, 더 가치 있는 서비스를 준비하겠습니다.
AI 신약개발 플랫폼 하이퍼랩
- 무료체험 : https://buly.kr/6ihK6EW
- 도입겁토 : https://abit.ly/yyytsg