ICML 2025 미리보기


2025년의 ICML
해마다 여름이면 3대 AI 학회 중 하나인 ICML 시즌이 다가옵니다. 올해에는 7월 13일부터 19일까지 The 42nd International Conference on Machine Learning(ICML 2025)의 본학회가 예정되어 있습니다. 글을 쓰고 있는 지금, 발표가 승인된 논문들의 camera-ready 버전(모든 수정이 완료되고 정제된 출판용 원고)이 제출되기를 앞두고 있는데요, 그에 앞서 일찍이 승인 논문들의 목록은 공개되었기에 이번에도 우리 분야에 관련된 논문들을 살펴보기로 했습니다. AI 학회 미리보기는 저희 AI 연구팀에서 돌아가며 맡고 있는데, ICML은 공교롭게도 작년에 이어 다시 제가 맡았네요 😄
ICML 논문들의 통계부터 살펴보겠습니다. AI는 반짝 유행이 아니라 트렌드라는 점에 모두가 공감할 정도로, 다양한 산업군과 정부 정책에까지 여전히 강세가 이어지고 있지요. 이는 ICML과 같은 AI 학회들에 투고되고 발표되는 연구 수에도 그대로 드러나고 있습니다. ICML에 투고된 논문 수는 근 2년간 부쩍 늘었는데요, 총 투고 수가 작년에는 9,653건, 올해에는 12,107건으로 무려 25%나 증가했습니다. [Paper Copilot].
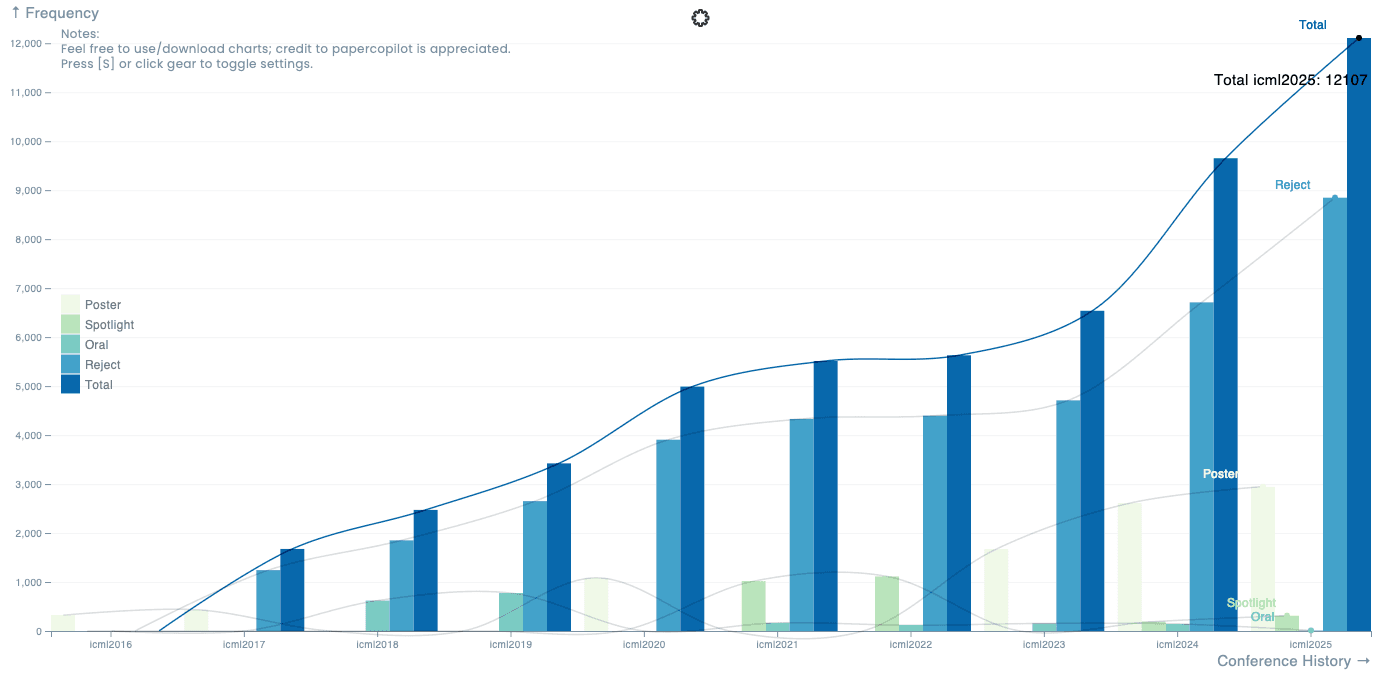
그런 반면, 포스터 발표, Spotlight, 구두 발표를 모두 포함해 발표가 승인된 논문 수는 3,260건(ICML 사이트에 따르면 3,340건)으로, 승인률은 27%였습니다. 작년 승인률이 31%였던 것에 비하면 다소 낮아졌네요. 점차 더 많은 연구가 이루어지고 있지만, 학회지에 게재되는 비율은 일정하게 조절되고 있는 것 같습니다. 승인률은 다른 유명 AI 학회들도 AAAI 2025의 경우 23%, ICLR 2025는 32%, NeurIPS 2024는 26% 등 다소간 차이는 있지만 비슷한 편입니다.

ICML 2025: 신약개발과 관련된 논문들
이번 미리보기에도 학회에 발표될 논문들 중 신약개발과 관련된 논문들을 먼저 모아보았습니다. 관련 논문들은 공식 논문 목록 페이지에서 chem, drug, mol 등 관련 키워드들을 검색하여 수집했습니다. 작년 ICML 2024 미리보기에서 모았던 관련 논문들은 60건이 조금 안 됐었는데요, 이번에는 그 건수가 100건을 넘어섰습니다. 이는 전체 논문 수가 증가한 것뿐만 아니라, (자연)과학 문제에 AI를 적용하는, 소위 AI4Science에 대한 AI 커뮤니티의 관심이 높아지고 있음을 반영하는 것으로 보입니다.
큐레이션한 논문 목록은 아래에서 확인할 수 있습니다. 신약 개발과의 관련성이 다소 느슨한 계산화학이나 재료 분야의 논문들도 일부 포함되어 있으니 참고해주시길 바랍니다 😉
ICML 2025 Papers for Drug Discovery
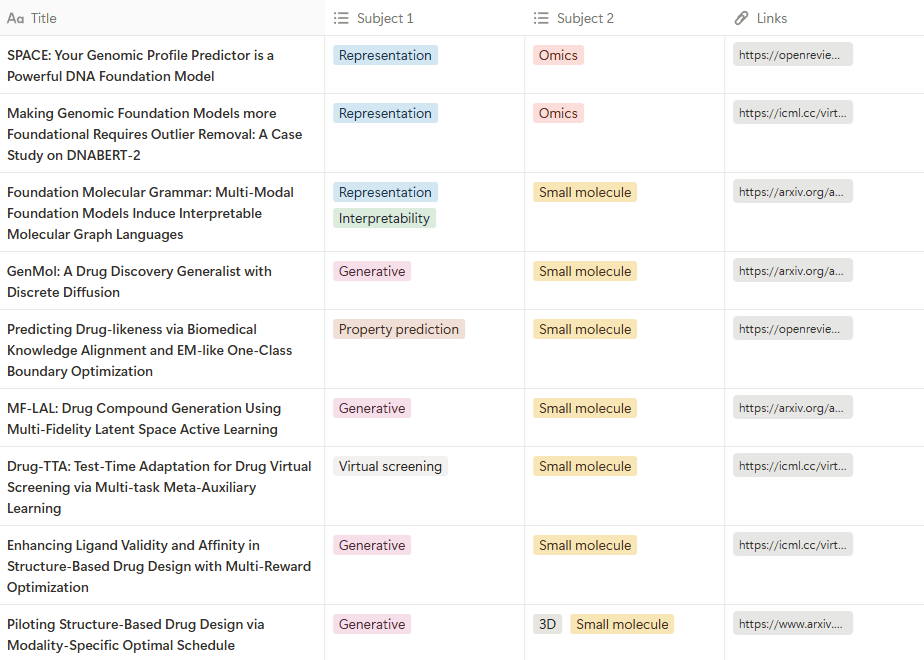
관련 논문 수가 전체적으로 증가하면서, 다뤄지는 분야도 더욱 다양해졌습니다. 소형 리간드나 단백질을 디자인하기 위한 생성모델 연구들이 여전히 두드러지는 반면, 다양한 오믹스 데이터를 다루는 생물정보학 주제도 눈에 띄게 늘어난 모습입니다. 또한 단백질과 같은 분자와 달리, 대칭성과 주기성이라는 독특한 특징을 지닌 결정(crystalline) 구조를 위한 생성모델 연구도 부쩍 증가했습니다.
다음으로는 아래의 논문들을 대표로 삼아, 이번 ICML 2025에서 발표될 연구들을 간략히 살펴보겠습니다.
- Shen, Seo et al. Compositional flows for 3D molecule and synthesis pathway co-design [ICML 2025].
- Wu, Padia et al. Identifying biological perturbation targets through causal differential networks [ICML 2025].
- Bendidi, El Mesbahi et al. A cross modal knowledge distillation & data augmentation recipe for improving transcriptomics representations through morphological features [ICML 2025].
- Joshi, Fu et al. All-atom diffusion transformers [ICML 2025].
ICML 2025 미리보기 1 | 3DSynthFlow: 3차원 분자 구조와 합성경로의 co-design
첫 번째로 살펴볼 논문은 Tony Shen, Seonghwan Seo 등 저자진의 “Compositional Flows for 3D Molecule and Synthesis Pathway Co-Design”입니다. 생성 AI를 이용한 분자 디자인에서는 흔히 분자의 3차원 구조(타깃 단백질에 대한 결합 구조)나 합성 가능성 중 한쪽에만 초점을 맞추는 경우가 많아, 다른 한쪽에 대한 성능이 떨어지는 문제가 발생하곤 합니다.
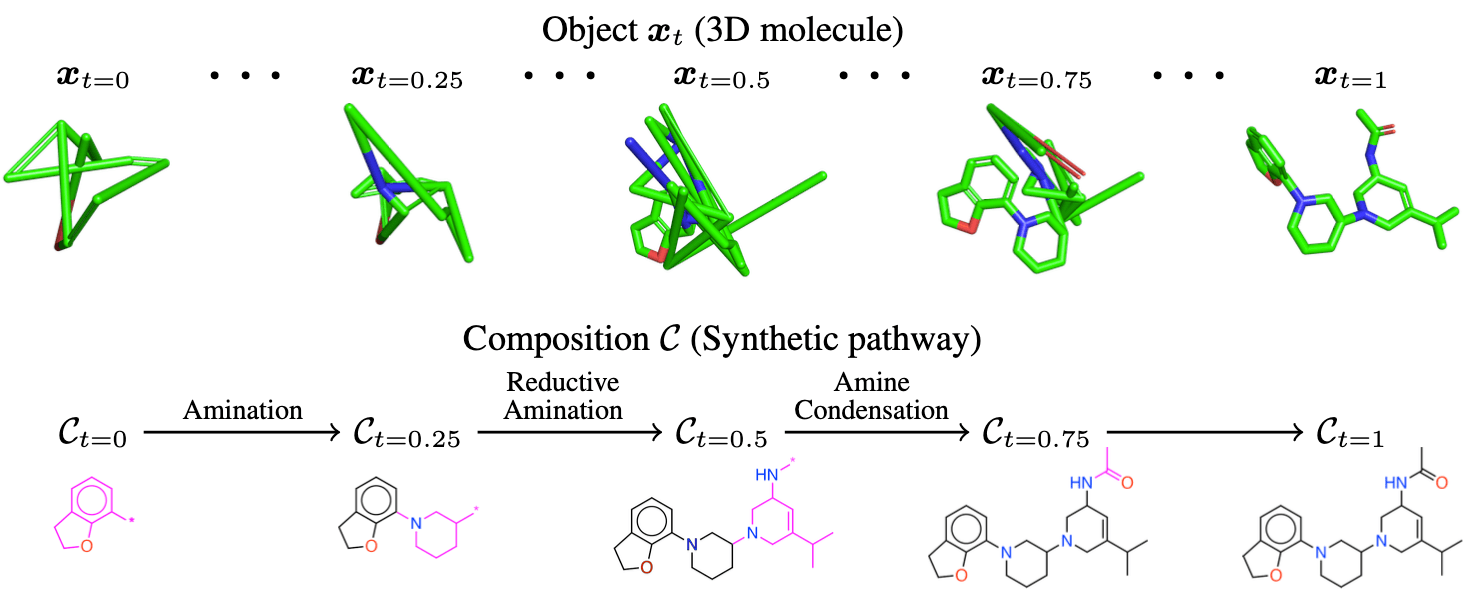
본 논문에서는 두 문제를 동시에 해결하기 위해 분자를 그 구성(composition)과 상태(state)로 표현합니다. 이때 분자의 구성이란 해당 분자 구조를 어떤 조각들이 어떻게 이어져 이루어져 있는지를 말하며, 실제 합성 과정에서 반응에 따라 부분 구조들이 조합되어 최종 분자를 구성하는 점을 묘사합니다. 상태란 분자의 3차원 구조, 즉 원자들의 3차원 좌표를 의미합니다. 논문에서는 생성 모델의 방법 중 하나인 flow matching을 이용하여 분자의 구성과 상태를 동시에, 상호의존적으로 생성하는 CGFlow(Compositional Generative Flow)를 제안하고, 이를 합성 가능한 리간드 디자인에 특화한 3DSynthFlow 모델을 소개합니다[1].
3DSynthFlow가 분자를 생성하는 과정은 아래 그림과 같습니다. 한 번의 생성 과정에서, 분자의 2차원 구조(즉, 구성)에서는 일정한 간격의 시점에 조각이 추가됩니다. 2차원 구조에서 조각이 추가될 때, 3차원 구조(즉, 상태)에서도 해당 조각에 대응하는 원자들이 함께 추가되는데요, 2차원 구조와 달리 3차원 구조는 매 연속된 시점에서 더 정제된 구조를 가지도록 갱신됩니다. 생성 과정이 모두 끝나면, 2차원과 3차원에서 모두 정제된 분자가 완성됩니다.
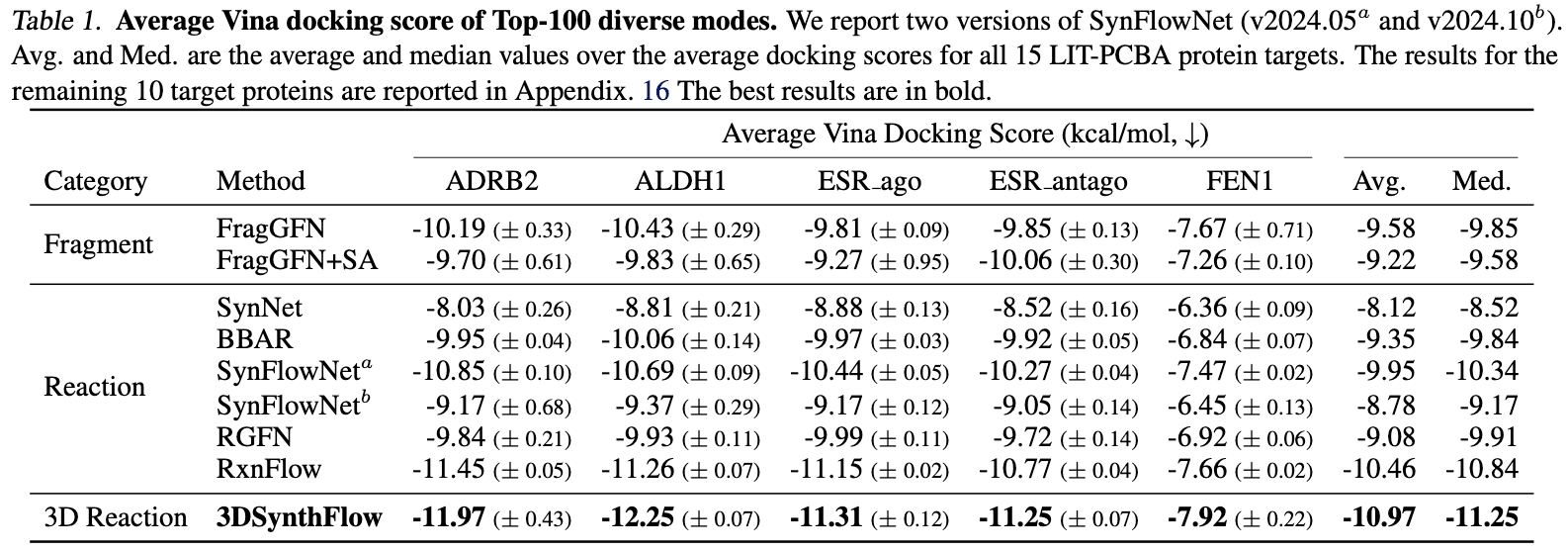
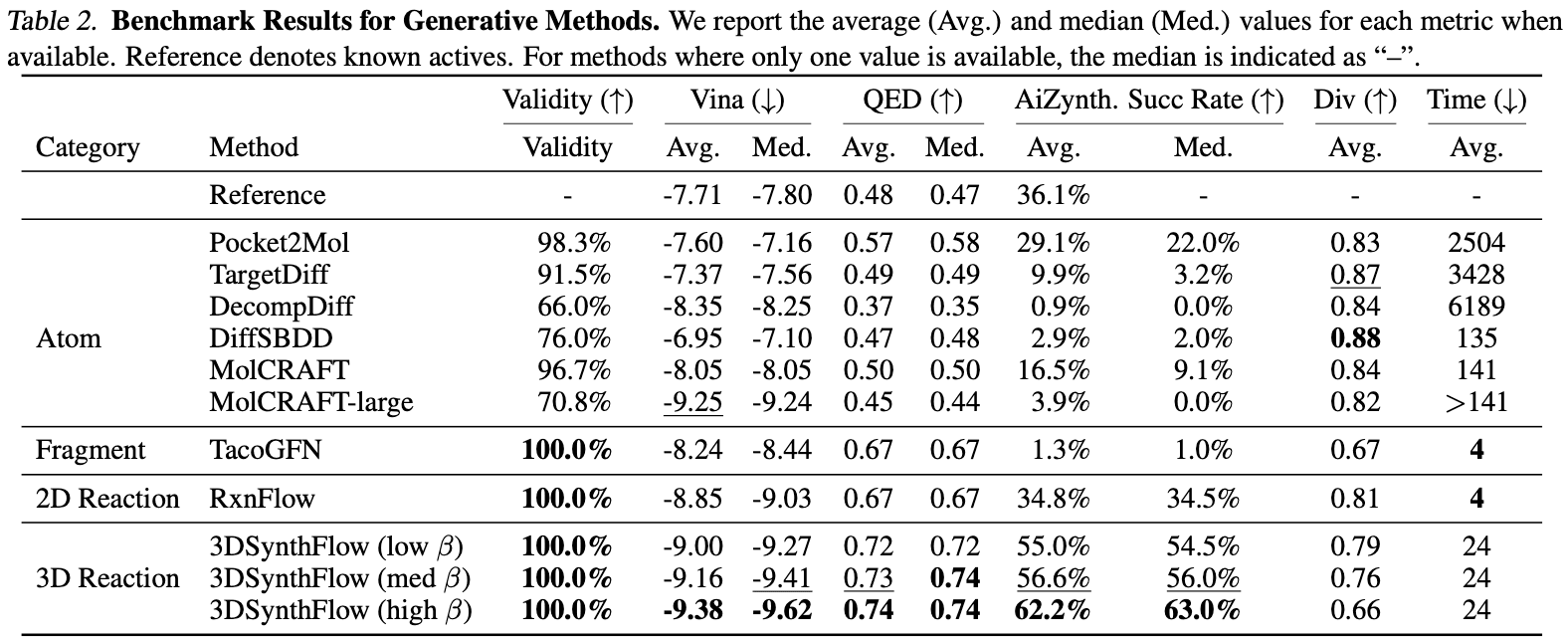
저자들은 3DSynthFlow를 통해 두 가지 성능을 입증했습니다: (1) 단일 타깃에 대한 활성을 최적화하는 성능과 (2) 단일 모델로 다양한(새로운) 타깃에 맞는 리간드를 생성하는 성능입니다. (1)에서는 다음 표와 같이 유사한 모델들에 비해 더 최적화된 도킹 점수를 얻는 리간드를 생성할 수 있었고,
(2)에서는 100가지 다양한 타깃에 대해 도킹 점수와 합성가능성 면에서 동시에 가장 우수한 성능을 보였습니다. 합성가능성은 AiZynthFinder \[Genheden et al. 2020]를 사용해 평가하였습니다.
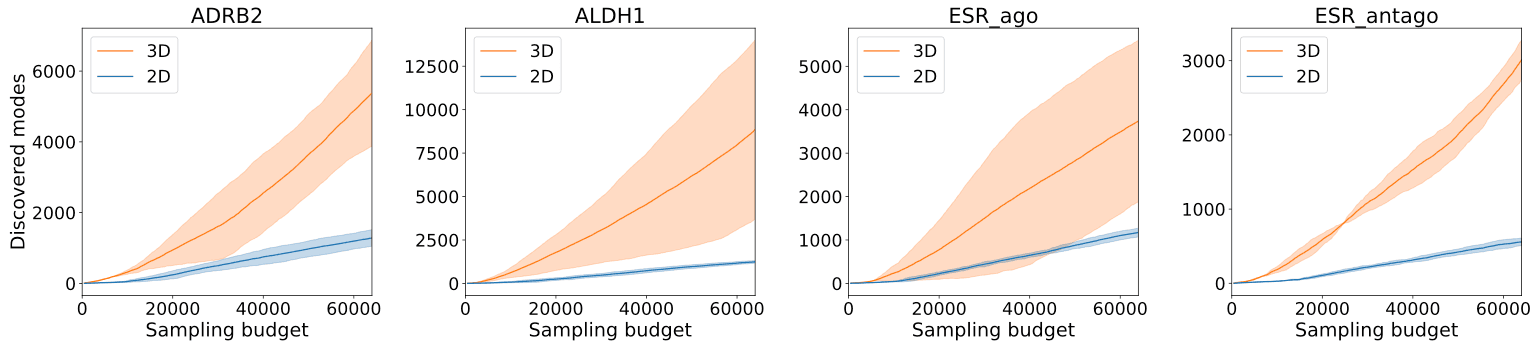
또한 3DSynthFlow는 분자를 2D 구조로만 다룬 저자들의 이전 모델에 비해, 3D 구조 정보를 함께 다룸으로써 단위 시간에 더 많은 분자 구조를 발굴할 수 있었습니다.
ICML 2025 미리보기 2 | CDN: 인과 네트워크를 통한 타깃 발굴
타깃 발굴은 신약개발의 시작이 되는 가장 중요한 단계라고 할 수 있습니다. 특정 질병을 치료하기 위해 공략해야 할 타깃, 혹은 특정 약이 작용하는 근본적인 타깃을 제대로 판명하지 못하면 뒤이은 연구가 모두 수포로 돌아가거나, 시판된 약의 부작용이 한참 후에야 밝혀질 수도 있죠.
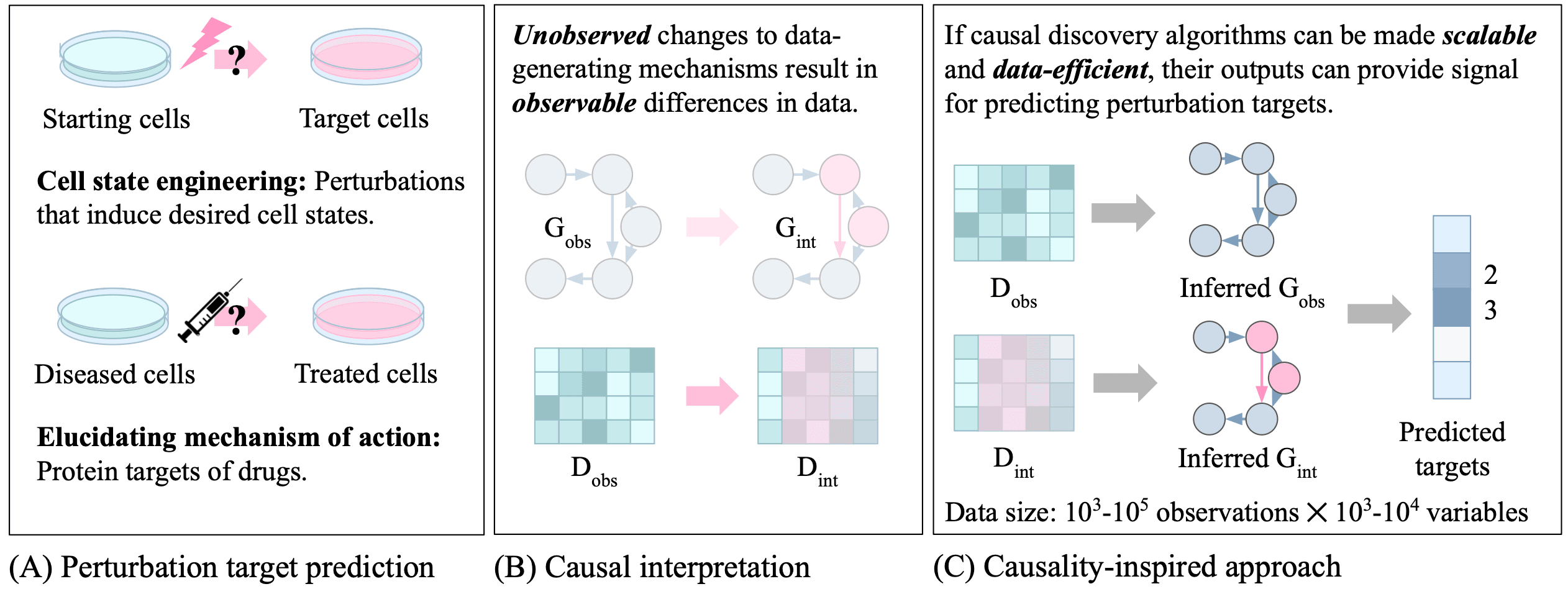
Menghua Wu, Umesh Padia 등 저자진의 “Identifying Biological Perturbation Targets through Causal Differential Networks”는 특정 섭동(perturbation 혹은 intervention)의 근본 원인이 되는 타깃 유전자를 예측하는 딥러닝 모델을 소개합니다. 논문의 모델인 CDN(causal differential networks)은 동일한 세포주에 섭동을 가하기 전의 전사체 데이터셋과 섭동을 가한 후의 전사체 데이터셋을 필요로 합니다. 각 데이터셋으로부터 먼저 해당 데이터셋을 설명하는(즉, 생성하는) 인과 그래프를 추정합니다. 인과 그래프는 꼭짓점(node)으로 유전자를 표현하고, 변(edge)으로 유전자 간의 관계를 표현합니다. 이렇게 얻은 섭동 전 인과 그래프와 섭동 후 인과 그래프의 차이를 분석함으로써, 최종적으로 해당 섭동의 타깃이 되는 유전자들을 예측하게 됩니다. 다시 말해, CDN은 기저의 인과관계 변화가 섭동 전후 관찰된 데이터 변화의 원인이라는 원리로 작동합니다.
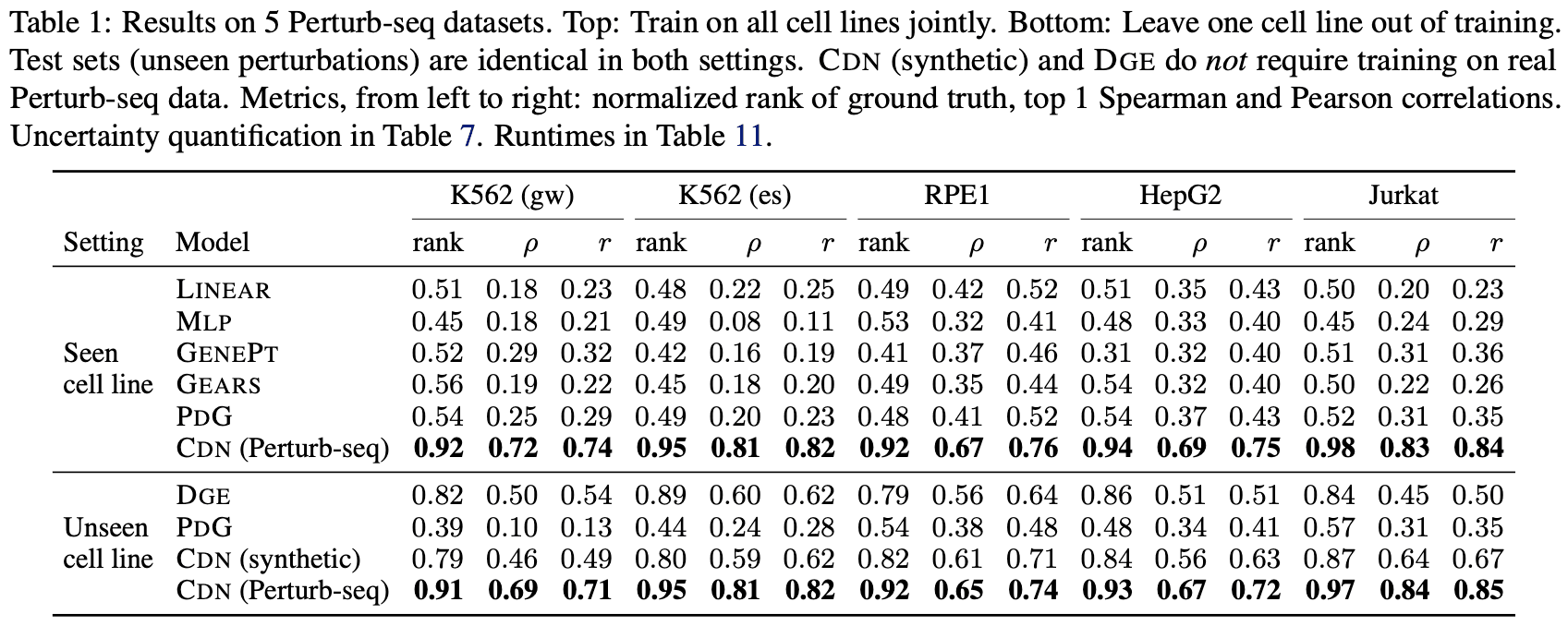
CDN은 두 가지 전사체 데이터에 대한 타깃 예측 성능을 평가했습니다. 먼저 CRISPR에 의한 Perturb-seq 데이터[2]를 사용해 평가했는데요, 아래 표에 나온 것처럼 기존 대조 방법들보다 월등히 우수한 예측 성능을 보여주었습니다. 이때 사용된 평가 데이터는 해당 유전자 섭동이 학습 데이터에 포함되지 않도록 분리되었으며, CDN은 여기에 더해 세포주 기준까지 추가로 분리했을 때에도 성능을 안정적으로 유지했습니다(아래 표의 “Unseen Cell Line”).
두 번째 평가는 약물로 유전자 발현에 영향을 가한 sci-Plex 데이터[3]를 사용해 진행되었습니다. 약물에 의한 섭동은 CRISPR보다 훨씬 간접적으로 유전자에 영향을 미치기 때문에, 기저 타깃을 판별하는 일이 훨씬 더 어렵습니다. 이러한 시나리오에서 CDN은 두 가지 세포주에 대한 평가에서 6가지 약물 중 3가지의 타깃을 정확히 예측하는 성과를 보였습니다. 다만 Infigratinib과 Palbociclib 같은 일부 약물에서는 성능이 크게 떨어졌는데요, 이를 통해 다양한 세포주와 약물 조합에 대해 타깃을 발굴하는 일은 여전히 상당한 도전 과제임을 알 수 있습니다.
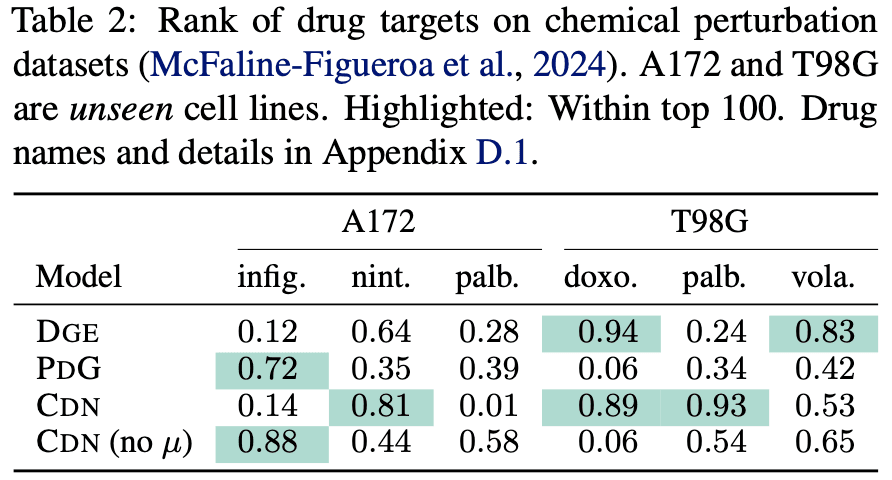
CDN은 입력 전사체 데이터 이외에 다른 외부 생물학 데이터에 의존하지 않는다는 점, 그리고 경쟁 모델 대비 연산 속도가 매우 빠르다는 점 등 여러 장점을 갖고 있습니다. 반면, 모델이 중간 표현으로 예측하는 인과 그래프가 생물학적 의미나 해석성을 지니는지에 대해 논문에서 별도의 분석이 이루어지지 않은 점은 다소 아쉬운 부분입니다.
ICML 2025 미리보기 3 | Semi-Clipped & PEA: 세포의 형태적 특성과 유전적 특성 연결짓기
세포의 기작을 연구하기 위한 대표적인 데이터 모달리티로는 전사체 데이터와 현미경 이미지 데이터가 있습니다. 전사체 데이터는 세포에 발현된 유전자들의 종류와 양을 나타내며, 이미지 데이터는 세포의 형태적 특징을 표현합니다. 동일한 세포의 전사체 데이터와 이미지 데이터를 함께 사용하면, 더욱 다양한 세포의 상황을 설명할 수 있는 일반화된 패턴을 학습할 수 있고, 이를 바탕으로 세포의 특성을 더 정확하게 예측할 수 있지 않을까요?
Recursion,Valence Labs, École Normale Supérieure – PSL(파리 고등사범학교) 소속의 Ihab Bendidi, Yassir El Mesbahi 등 저자진의 “A Cross-Modal Knowledge Distillation & Data Augmentation Recipe for Improving Transcriptomics Representations through Morphological Features”에서는 OpenAI의 유명한 CLIP(Contrastive Language–Image Pre-training) 방법을 응용하여 위 질문의 답을 제안합니다.
CLIP은 본래 이미지와 그에 대응되는 텍스트를 서로 연결짓도록 딥러닝 모델을 학습하기 위해 고안된 방법입니다. 기본 원리는 아래 그림에 나온 것처럼, 이미지의 임베딩과 텍스트의 임베딩을 동일한 공간에서 정의하되, 서로 연관된 이미지와 텍스트는 임베딩 간 유사도가 높아지도록 학습하고, 반대로 연관되지 않은 이미지–텍스트 쌍은 임베딩 간 유사도가 낮아지도록 학습하는 방식입니다.

본 논문에서는 세포의 현미경 이미지를 선생(teacher) 모달리티, 전사체를 학생(student) 모달리티로 정의하고, CLIP 메커니즘을 통해 학생이 선생의 특징을 배우도록 학습시킵니다. 이때 차이점은 이미지 임베딩은 고정된 상태로 유지되고, 전사체 임베딩만 CLIP의 목적함수에 의해 학습된다는 점입니다. 이러한 방식으로 인해 저자들은 본 방법을 Semi-Clipped라 명명합니다. 이렇게 semi-clipping을 적용하면, 학습이 끝난 후 (1) 결과적으로 하나의 모달리티인 전사체만을 사용한 예측이 가능해지고, 동시에 (2) 이미지 데이터에 담긴 지식을 전사체 데이터에 증류(knowledge distillation)함으로써, 전사체만으로 학습했을 때보다 예측 정확도를 높일 수 있습니다.
논문에서는 위와 같은 지식 증류와 함께, 세포 실험 데이터가 고질적으로 겪는 배치 효과(batch effect)를 데이터 증폭의 형태로 해결합니다. 배치 효과란 동일한 실험을 반복 수행할 때 환경, 시각, 실험자 등의 다양한 변인에 의해 실험 결과가 달라지는 문제를 말합니다. 논문에서는 전사체 데이터에 대해 미리 정의된 정규화 방법들 중 하나를 학습 중 무작위로 적용하고, 이미지 데이터에는 항상 동일한 정규화를 매번 적용합니다. 이렇게 정규화된 전사체 임베딩과 이미지 임베딩을 기반으로 CLIP 목적함수를 계산하고, 이를 통해 전사체 임베딩 네트워크가 지식 증류를 위한 방향으로 학습됩니다.
이러한 무작위성은 학습 데이터를 증폭시키는 효과도 가져오는데요, 이는 실제로 서로 연관된 전사체–이미지 데이터 쌍이 부족한 상황을 보완하는 데 유용합니다. 특히 동일한 세포로부터 전사체 데이터와 이미지 데이터를 동시에 얻는 것은 실험적으로 매우 어렵기 때문에, 논문에서는 두 데이터의 연관성을 세포주, 섭동(perturbation) 조건 등의 메타정보 수준에서만 고려합니다. 이러한 데이터 증폭 기법은 PEA(perturbation embedding augmentation)라 명명되었습니다.
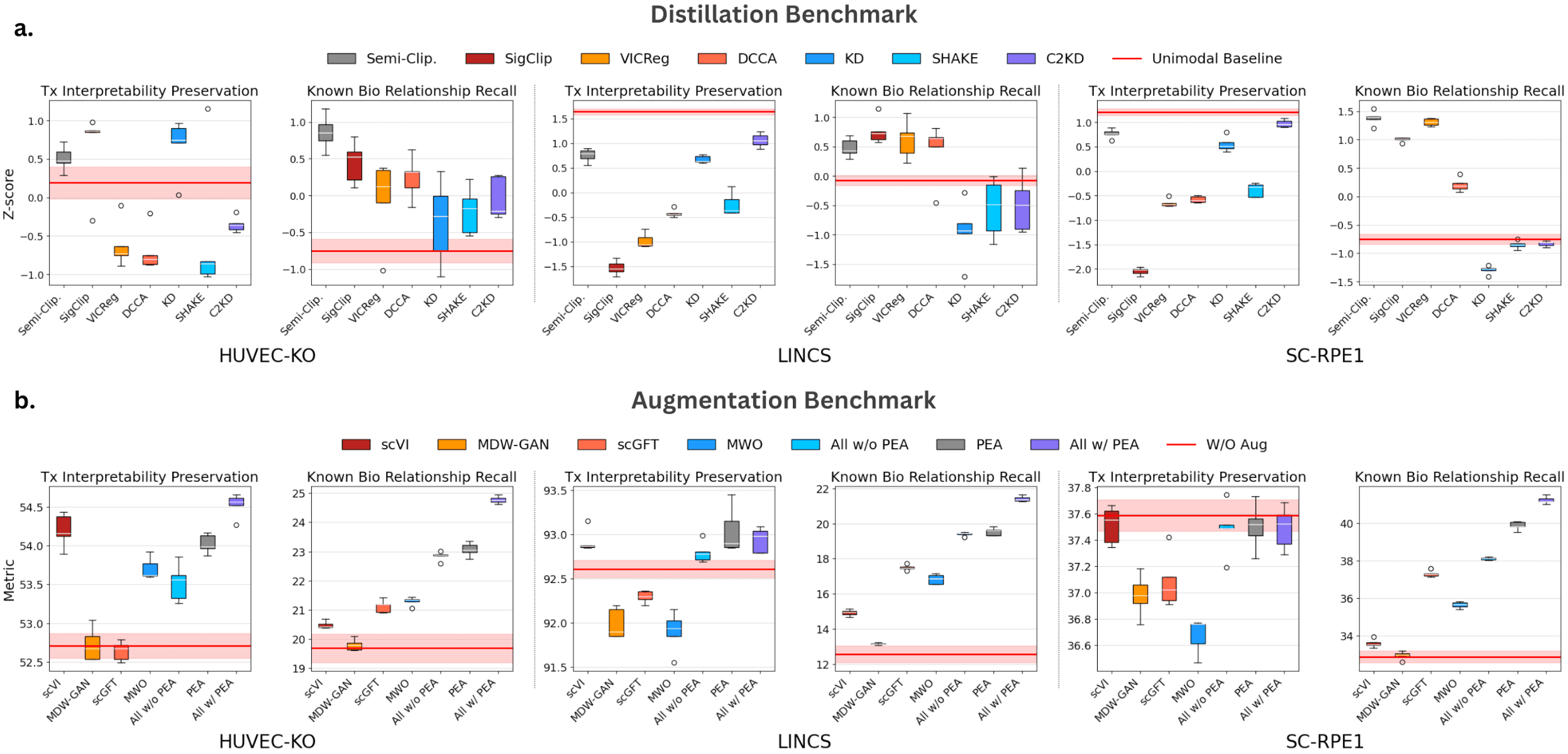
논문에서는 제안한 증류 방법과 증폭 방법을 세 가지 분포 외(out-of-distribution) 데이터셋을 이용해 평가합니다. 각 방법에 대한 평가는 두 가지 측면에서 이루어졌는데요, 첫 번째는 전사체 해석 보존력(transcriptomic interpretability preservation)으로, 이는 지식 증류를 위한 변환을 거친 전사체 임베딩이 원본 전사체 데이터의 기본적인 정보를 얼마나 잘 유지하는지를 측정합니다. 두 번째는 알려진 생물학적 관계 추출력(known biological relationships retrieval)으로, 학습된 임베딩이 이미 알려진 유전자 간 관계를 얼마나 잘 포착하는지를 평가합니다. 아래 그래프에서 볼 수 있듯, 두 지표 모두에서 수치가 클수록 성능이 좋은데요, 두 측면 모두에서 Semi-Clipped와 PEA가 기존 대조 방법들보다 뛰어난 성능을 보이고 있습니다.특히 섭동 방식(화학적 섭동 vs CRISPR), assay 기법, bulk 세포 vs 단일 세포 등 다양한 조건을 고려한 평가 데이터셋을 구성해, 제안한 방법의 일반화 성능을 검증한 점이 인상 깊습니다.
ICML 2025 미리보기 4 | ADiT: 분자와 재료 시스템을 아우르는 파운데이션 모델
분자의 3차원 구조를 생성하는 생성 AI의 발전이 거듭되면서, 최근에는 저분자부터 단백질 같은 고분자에 이르기까지 여러 스케일의 분자 시스템을 한 번에 다룰 수 있는 모델이 주목받고 있습니다. Meta의 FAIR(Fundamental AI Research)에서 발표할 “All-atom Diffusion Transformers”에서는 약간 다른 접근을 통해, 결정 시스템과 비결정 분자 시스템을 하나의 모델로 생성하는 방법을 제안합니다.
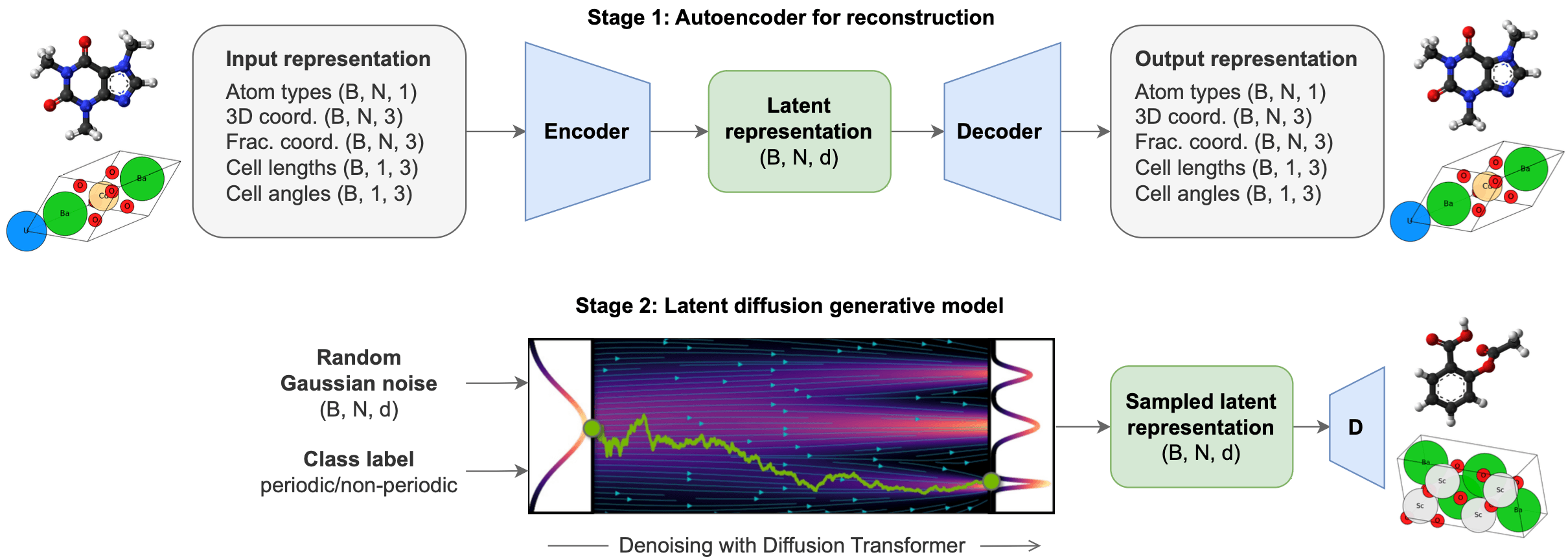
본 논문의 프레임워크인 ADiT(All-atom Diffusion Transformer)는 먼저 Variational Autoencoder(VAE)를 이용해 결정 구조와 비결정 구조가 동시에 임베딩된 잠재 공간(latent space)을 학습합니다. 이때 VAE의 디코더는 해당 임베딩으로부터 3차원 결정 구조 또는 비결정 분자 구조를 복원할 수 있도록 훈련됩니다.
그다음, 새로운 구조를 생성할 수 있도록 DiT(Diffusion Transformer, 확산 트랜스포머)가 활용됩니다. 3차원 분자 구조 생성에서 이미 뛰어난 성능을 보이고 있는 확산 모델의 원리를 ADiT는 VAE의 잠재 공간에 적용하는데, 이를 latent diffusion이라 부릅니다. 새로운 구조를 생성할 때는 먼저 표준 정규분포로부터 무작위로 임베딩을 샘플링한 뒤, DiT를 통해 denoising 과정을 거칩니다. 이후, 이 denoising된 임베딩을 VAE의 디코더가 실공간의 분자 구조로 복원하게 됩니다.
아래 그림은 위 같은 ADiT의 all-atom 임베딩과 DiT 과정을 표현합니다.
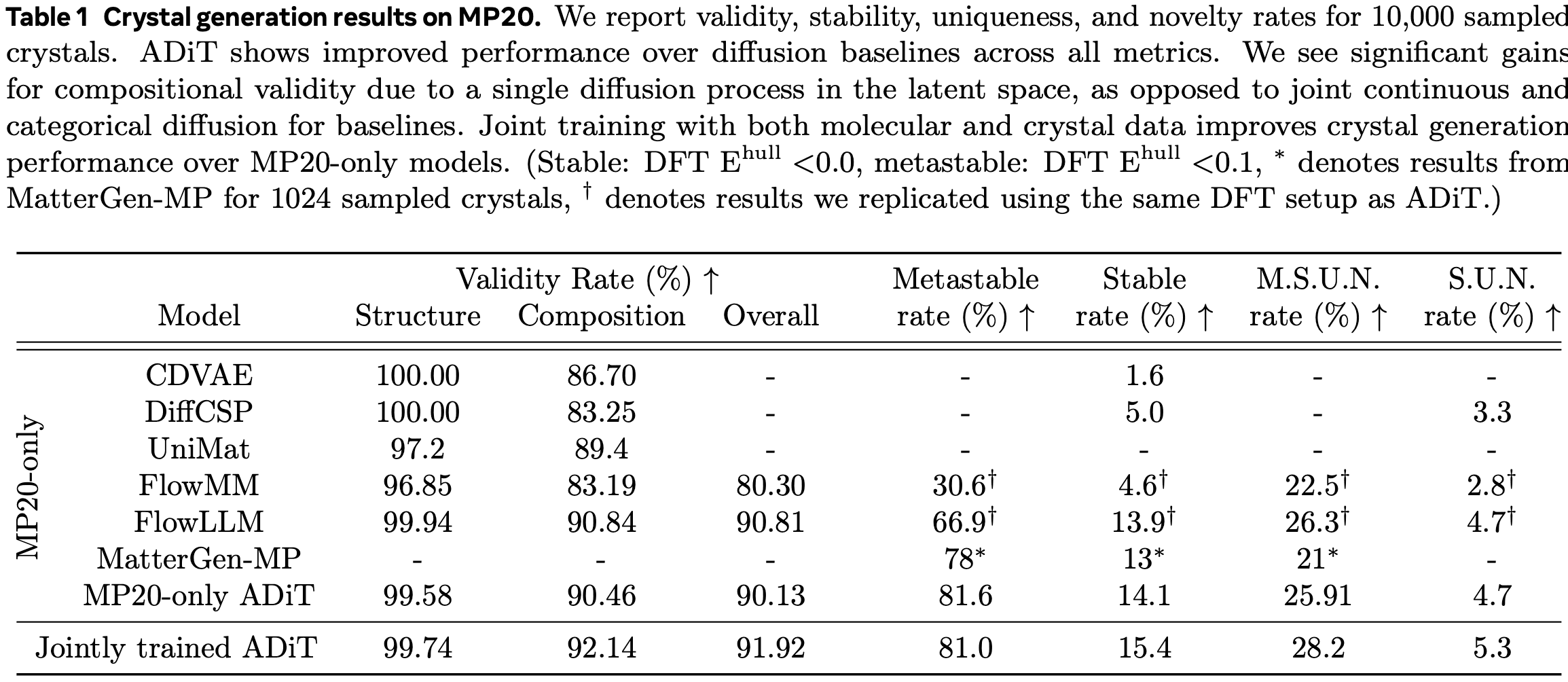
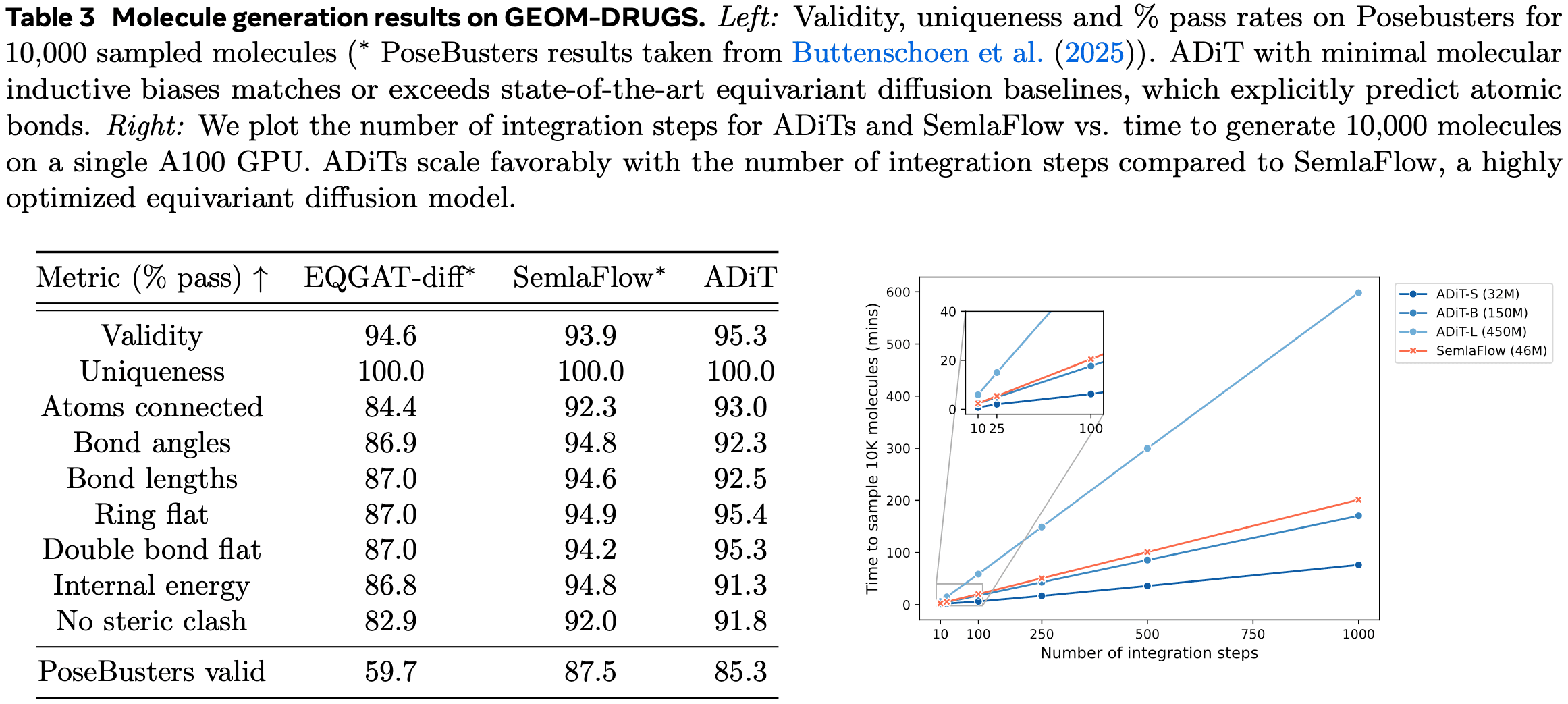
결정 시스템과 비결정 시스템을 동시에 다룰 수 있다는 점을 강점으로 내세우는 만큼, ADiT의 생성 성능은 각 시스템에 대해 따로 평가되었습니다. 먼저, 아래 표에서 볼 수 있듯 결정 구조 생성에 있어서 ADiT는 state-of-the-art 성능을 보여주었습니다. 또한 결정 데이터와 비결정 분자 데이터를 함께 학습할 경우 성능이 개선되는 경향도 확인되었는데요, 그 향상 폭이 아주 크다고 보기는 어렵습니다.
다음으로는 비결정 분자 구조의 생성 성능을 살펴보았습니다. ADiT의 또 다른 특징은 3차원 구조 생성에서 흔히 사용되는 등변성(equivariance)을 구조적으로 포함하지 않았다는 점입니다 \[4]. 대신, ADiT는 학습 데이터의 증폭을 통해 등변성 부재를 보완합니다. 아래 표에서 비교된 대조 모델인 EQGAT-diff와 SemlaFlow는 모두 등변성을 보장하는 아키텍처를 기반으로 합니다. 반면 ADiT는 등변성을 구조적으로 보장하지 않음에도 불구하고, 성능이 더 뛰어나거나 유사한 수준을 유지하면서도 연산량이 훨씬 작습니다. 이러한 효율성 덕분에 ADiT는 모델의 크기를 보다 쉽게 확장하여 더 많은 데이터를 학습시킬 수 있는 장점이 있습니다.
마지막으로는 시각적 예시로 ADiT가 금속 유기 골격체 (metal–organic frameworks, MOFs)를 생성한 예시들을 보여드립니다.
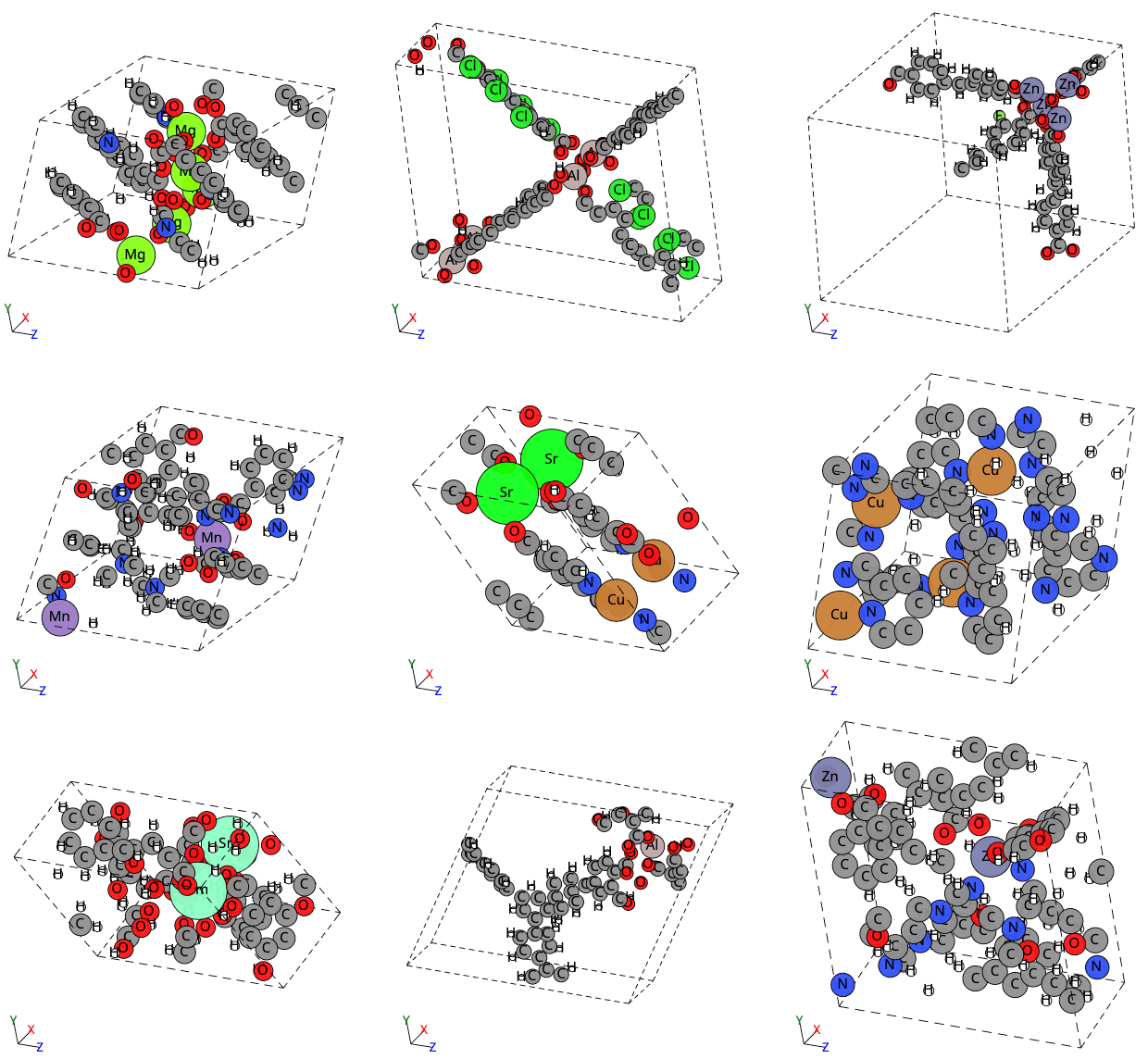
ADiT 논문의 가장 아쉬운 점은 시연된 시스템들이 비교적 작다는 것입니다. 평가에 사용된 시스템들은 원자 개수가 많아야 100개를 조금 넘는 수준인데, 단백질이나 핵산 같은 분자 시스템은 수백, 수천 개의 원자로 이루어져 있죠. 물론 저자들도 이 한계를 인식하고 있으며, ADiT의 높은 확장성(scalability)이 이러한 문제를 해결할 수 있는 가능성을 보여준다고 언급하고 있습니다.
조만간 AlphaFold 3처럼 다양한 스케일의 생체분자는 물론, 결정형 재료 구조까지 동시에 생성할 수 있는 진정한 파운데이션 AI도 등장하겠죠? 그런 통일된 하나의 AI 모델이 과연 꼭 남다른 장점을 가질지는 아직 단언할 수 없지만, 모든 원자 시스템의 기반이 되는 물리적 원리를 학습하는 데에는 분명 도움이 될 것 같습니다.
담지 못한 이야기
여건으로 인해 개별 논문은 위에서처럼 간단히만 살펴볼 수 있었는데요, 앞서 언급했듯 AI4Science에 대한 관심은 빠르게 증가하고 있으며, 그에 따라 흥미로운 연구들도 더욱 많이 등장하고 있습니다. 신약개발과 같은 문제에 거대 언어 모델(LLM)을 적용하는 논문들도 여전히 활발히 발표되고 있지만, 이번 미리보기에서는 생략했습니다. 실제로 LLM 응용 연구는 신약개발보다는 의학 분야에 적용된 경우가 특히 많습니다. ICML 2025 논문 목록 페이지에서 “medical”과 같은 키워드로 검색해보면, 관련된 논문들을 손쉽게 찾아볼 수 있습니다.
하반기의 NeurIPS 2025 미리보기에서는 더욱 재미있는 연구들을 살펴보도록 하겠습니다.
[1] 정확히는 CGFlow는 분자라는 특정 없이, 구성적 단계와 연속된 변수로 표현해야 하는 임의의 대상에 대해 적용할 수 있는 일반화된 방법으로 소개한다.
[2] Dixit et al. 2016; Replogle et al. 2022; Nadig et al. 2025.
[3] Srivatsan et al. 2019; McFaline-Figueroa et al. 2024.
[4] 분자 시스템과 등변성의 중요성을 이해하기에 좋은 글 [White 2021].
AI 신약개발 플랫폼 하이퍼랩
- 무료체험 : https://buly.kr/D3ebOOb
- 도입겁토 : https://abit.ly/yyytsg