Hyper Binding Co-folding: 알파폴드3 이후, 도킹 기술이 바꾸는 신약개발 패러다임


안녕하세요, 히츠의 CTO 임재창입니다.
최근 AI 기술의 눈부신 발전은 다양한 연구 분야에 새로운 가능성을 제시하고 있습니다. 특히 AlphaFold의 등장은 생명과학과 의약학 연구에 큰 전환점을 가져왔으며, 이제는 후속 버전인 AlphaFold3에 대한 관심도 높아지고 있습니다.
하지만 신약 개발이라는 복잡하고 정밀한 과정을 실제로 지원하기 위해서는, 단순한 단백질 구조 예측을 넘어, 보다 정교한 분자 간 상호작용 예측 기술이 필요합니다. 이에 히츠(HITS)에서는 최신 AI 기술을 접목해 신약 개발 연구자들이 실제 현장에서 체감할 수 있는 성능과 사용 편의성을 갖춘 Hyper Binding Co-folding 기술을 개발했습니다.
이번 글에서는 AlphaFold3의 기술적 진보와 이를 기반으로 한 Hyper Binding Co-folding 기술이 어떻게 신약 개발의 효율성과 정확성을 극대화할 수 있는지 자세히 소개해 드리겠습니다.
AlphaFold3와 분자 도킹: 신약 개발의 새로운 지평
2024년 Nature지를 통해 발표된 AlphaFold3는 단백질뿐만 아니라 DNA, RNA, 리간드 등 다양한 분자 간의 상호작용 예측에서 놀라운 성공률을 보여주며, 신약 개발 분야에 새로운 가능성을 제시했습니다. AlphaFold2까지는 주로 단백질 구조 예측이 주를 이뤘다면, AlphaFold3는 리간드 결합 예측(PoseBusters Set), 핵산과의 상호작용, 공유 결합 변형 등 다양한 영역에서 기존 도구들(예: AutoDock Vina, RoseTTAFold)을 능가하는 성능을 보여주었습니다. 실험 연구자들 사이에서 컴퓨터 기반 신약 개발(CADD) 소프트웨어의 사용이 점차 증가하는 추세이지만, 기존 도킹 방법들은 몇 가지 한계점을 가지고 있습니다. AlphaFold3의 이런 발전은 분자 도킹(Molecular Docking)의 한계점을 극복할 수 있는 혁신적인 솔루션으로 주목받고 있습니다.
기존 도킹 방법의 3가지 주요 한계점
- 단백질의 유동성 간과:
- 단백질은 고정된 구조가 아니라 끊임없이 움직이는 유동적인 분자입니다. 리간드가 결합할 때 단백질의 구조가 변할 수 있으며(유도 적합, Induced fit), 이러한 미세한 구조 변화도 결합 에너지 예측에 큰 영향을 미칠 수 있습니다.
- 대부분의 기존 도킹 소프트웨어는 단백질을 고정된 구조(Rigid body)로 간주하거나, 유동성을 고려하더라도(Flexible docking) 그 예측 정확도가 높지 않았습니다. 이는 단백질이 수천 개의 원자로 구성되어 계산 복잡도가 매우 높기 때문입니다.
- 분자 동역학 시뮬레이션을 활용하면 단백질의 유동성을 고려할 수 있지만, 일반적인 도킹 계산에 비해 수만 배나 느리고(도킹: 수 분 vs MD: 수 시간~수일), 전문 지식과 복잡한 설정이 필요하여 실험 연구자들이 직접 수행하기 어렵습니다.
- 결합 위치 예측의 어려움:
- 이론적으로 약물은 단백질 표면 어디에나 결합할 수 있지만, 모든 경우의 수를 탐색하는 것은 계산적으로 불가능에 가깝습니다. 따라서 일반적으로 사용자는 리간드가 결합할 것으로 예상되는 특정 영역(Binding site)을 지정해 주어야 합니다.
- 만약 결합 위치를 모르는 상태에서 전체 단백질을 대상으로 탐색하는 블라인드 도킹(Blind docking)을 수행하면, 계산 시간이 많이 증가하고 예측 정확도는 현저히 감소하는 경향이 있습니다.
- 입력 단백질 구조의 질에 따른 성능 편차:
- 도킹 시뮬레이션의 정확도는 입력으로 사용되는 단백질 3차원 구조의 질에 크게 좌우됩니다.
- 단백질 서열만 아는 경우: 기존 도킹 방법은 적용이 거의 불가능하며, 상동성 모델링(homology modeling)을 통해 구조를 예측해야 하지만 정확도가 낮을 수 있습니다.
- Apo 구조 (리간드가 결합되지 않은 구조)만 있는 경우: 도킹 성능이 감소할 수 있습니다. 대부분의 신규 타겟은 이 경우에 해당합니다.
- Holo 구조 (리간드가 결합된 구조)가 있는 경우: 가장 좋은 경우이지만, 항상 이용 가능한 것은 아닙니다.
- 특히 "First-in-class" 혁신 신약을 목표로 하는 신규 타겟일수록 실험적으로 규명된 3차원 구조, 특히 리간드 결합 구조(Holo 구조)가 없는 경우가 많아 어려움이 큽니다.
- 도킹 시뮬레이션의 정확도는 입력으로 사용되는 단백질 3차원 구조의 질에 크게 좌우됩니다.
기존의 한계를 극복한 Docking: Hyper Binding Co-folding
이러한 기존 도킹 기술의 한계점을 극복하기 위해, HITS는 AlphaFold3의 개념을 확장하여 Hyper Binding Co-folding이라는 새로운 도킹 기술을 개발했습니다. 이 기술은 단백질, RNA, DNA, 리간드의 서열 정보로부터 직접 복합체 구조를 예측하는 Co-folding 방식을 기반으로 합니다. Hyper Binding Co-folding은 다음과 같은 4가지 주요 장점을 제공합니다.
- Induced fit의 정확한 예측: 단백질 서열 정보만으로 각 리간드에 최적화된 단백질 구조(induced fit)를 제공하여 예측 정확도를 크게 향상시킵니다. 이는 리간드 결합 시 발생하는 단백질의 미세한 구조 변화를 반영하여, 리간드-단백질 상호작용 예측의 정확도를 높입니다.
- 결합 위치(Binding Site) 설정 불필요: 기존 도킹과 달리, 사용자가 결합 위치를 미리 지정해 주지 않아도 정확한 예측이 가능합니다. 물론, 결합 위치 정보를 제공하면 예측 정확도를 더욱 높일 수 있습니다.
- 단백질 3D 구조 준비 과정 간소화: 단백질 서열만 선택하면 되므로, 기존의 복잡했던 최적의 단백질 3D 구조 선택이 필요하지 않으며, 이 과정 없이도 높은 정확도를 얻을 수 있습니다.
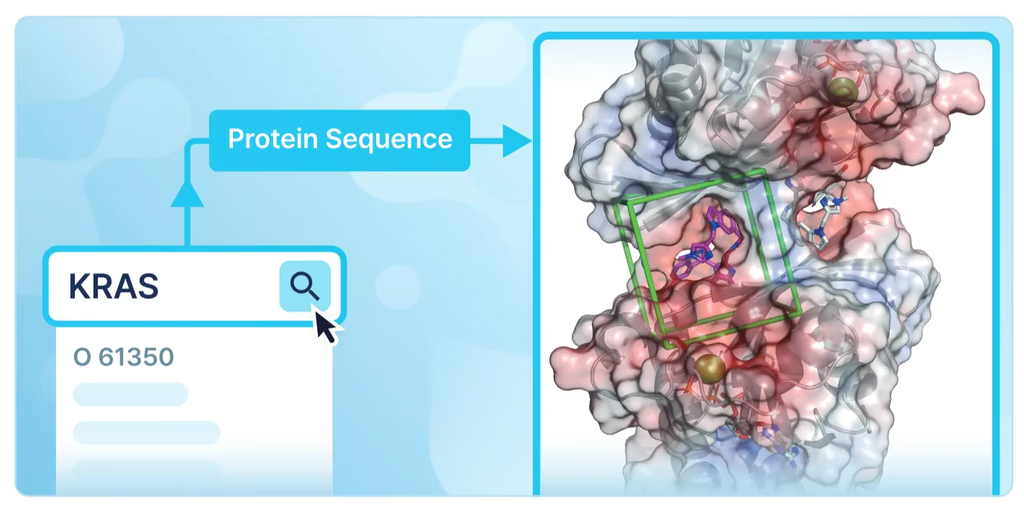
- 압도적인 사용자 편의성: 단백질 구조 기반 신약 개발에서 실험 연구자들이 가장 어려워하는 과정 중 하나인 입력 단백질 구조 설정을 단순화했습니다. 단백질 이름(예: KRAS)만 검색하면 관련된 정보(UniProt ID, 유전자명, 생물종 등)가 자동으로 제공되어 최적의 인풋 설정이 가능합니다.
사용자 워크플로우는 매우 간단합니다:
- 단백질 이름 입력 (HITS 데이터베이스 자동 검색)
- 단백질 서열, 보조 인자, 금속 이온, 번역 후 변형(PTM) 등 정보 확인 및 수정
- 리간드 2D 구조 입력 (리간드 컨포머 자동 생성)
- 리간드-결합 구조 및 결합 에너지 예측
Hyper Binding Co-folding의 성능 및 응용
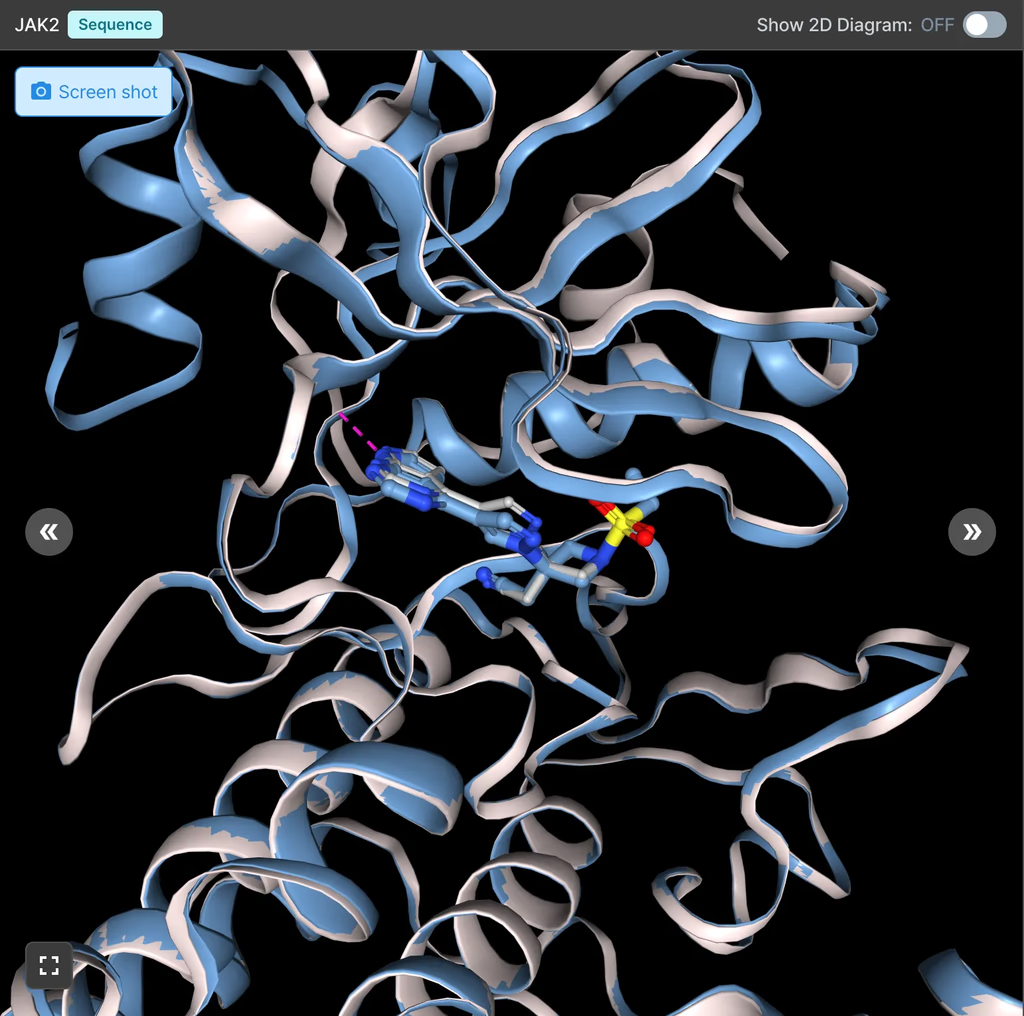
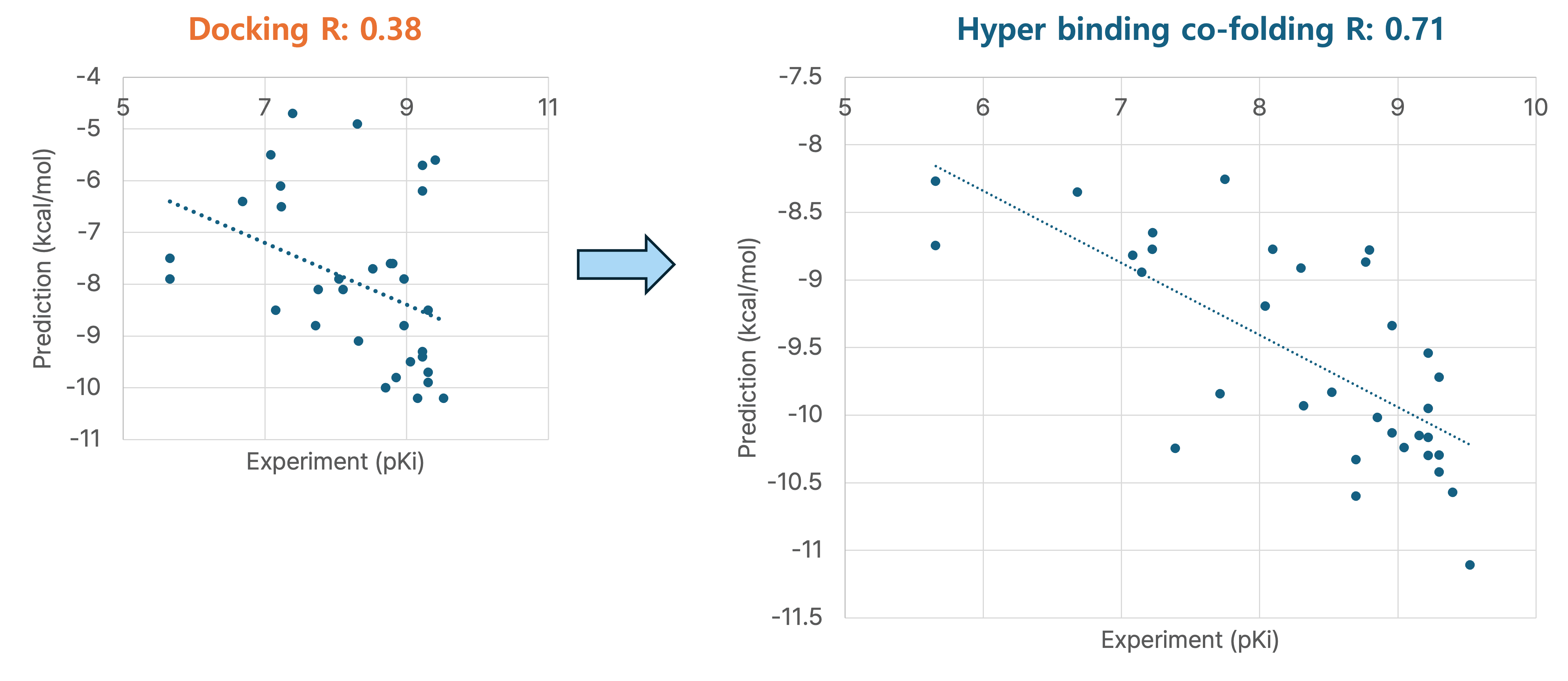
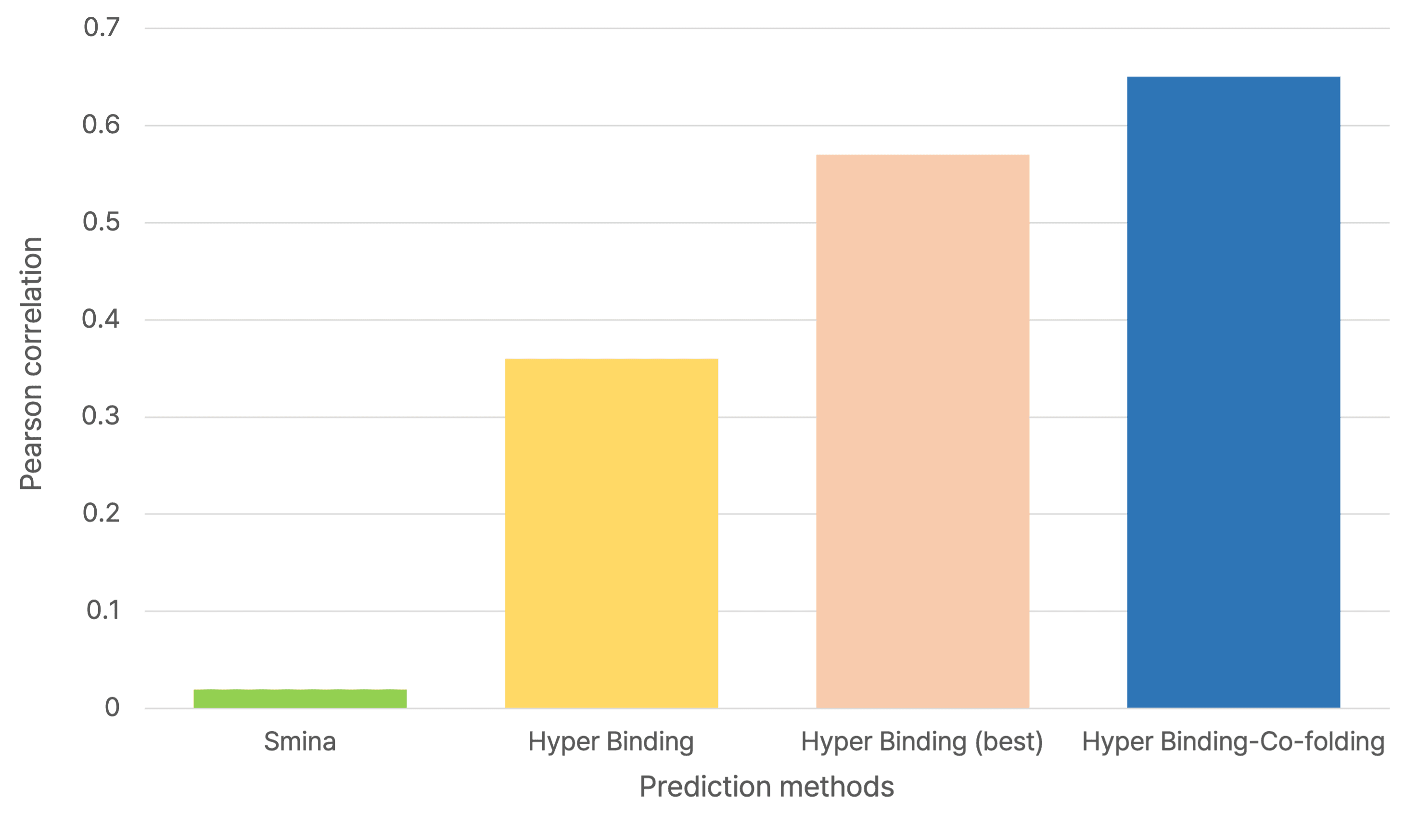
1. Janus Kinase 2 (JAK2) 저해제 결합 예측
JAK2 저해제에 대한 결합 에너지 예측에서, 기존의 Hyper Binding 방법은 실험값(pKi)과의 Pearson correlation coefficient (R) 값이 0.38이었으나, Hyper Binding Co-folding을 적용했을 때 R 값이 0.71로 크게 향상되었습니다. 이는 실제 실험값을 훨씬 더 정확하게 예측함을 의미합니다.
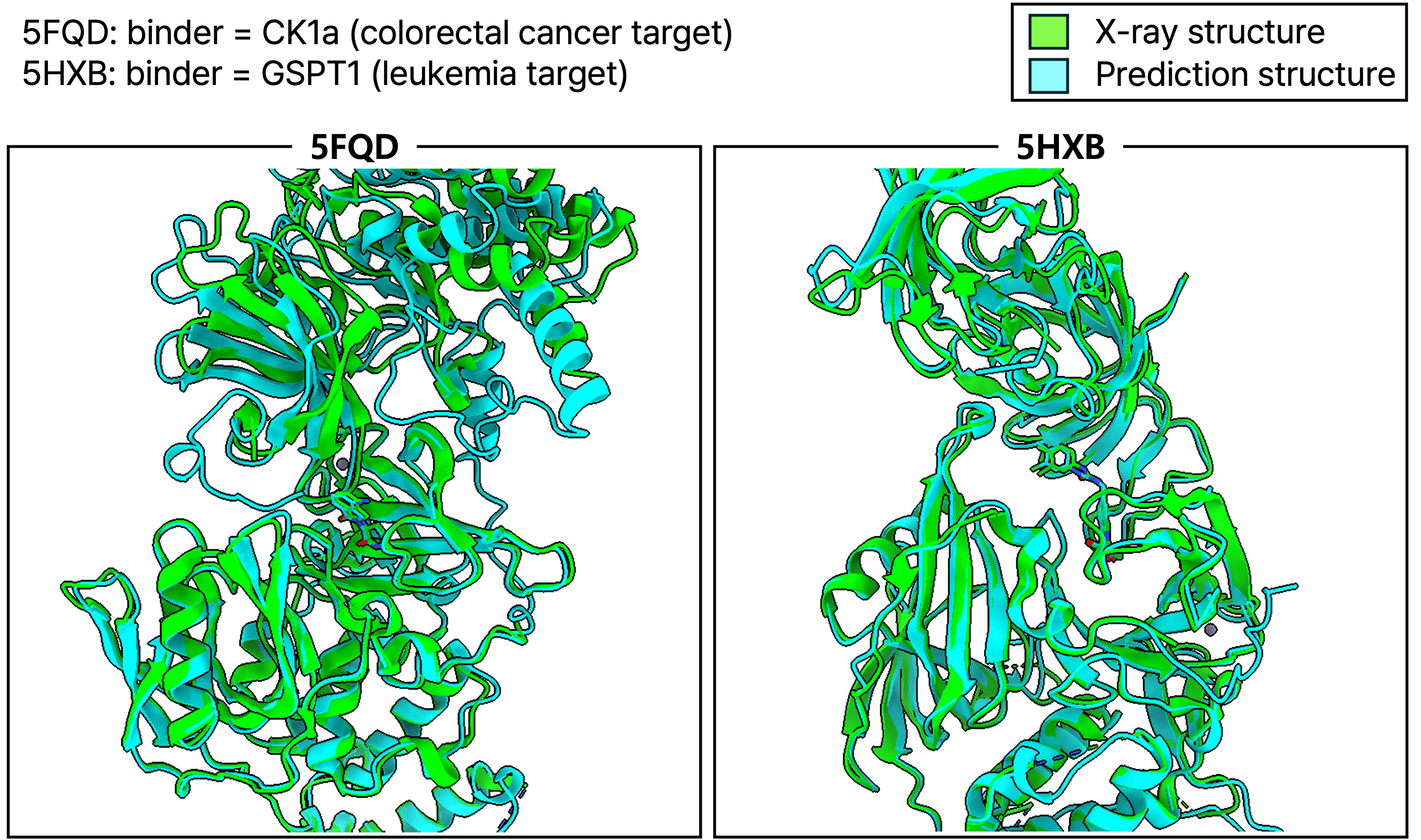
2. Molecular Glue 예측
Molecular glue는 두 단백질 사이의 상호작용을 유도하여 표적 단백질의 분해를 촉진하는 저분자 화합물입니다. 이는 기존에 "undruggable"로 여겨졌던 단백질 타겟을 공략할 수 있는 새로운 전략으로 주목받고 있습니다. Hyper Binding Co-folding은 이러한 복잡한 삼중 복합체(ternary complex) 구조 예측에도 성공적으로 적용될 수 있음을 보여주었습니다. 예를 들어, 대장암 타겟인 CK1a에 결합하는 molecular glue (PDB: 5FQD)와 백혈병 타겟인 GSPT1에 결합하는 molecular glue (PDB: 5HXB)의 경우, 예측 구조가 실제 실험 구조와 높은 유사성을 보였습니다. 특히 molecular glue 존재 유무에 따른 E3 ligase와 binder (GSPT1) 단백질의 구조 변화도 예측 가능했습니다.
3. 예측 정확도 비교
- 리간드-단백질 결합 구조 예측 정확도 (PoseBusters v2 기준): Hyper Binding Co-folding은 DiffDock, AutoDock Vina 등 기존 방법들 및 AlphaFold3보다 높은 예측 정확도를 보였으며, 특히 결합 위치 정보를 활용했을 때 더욱 향상된 성능을 나타냈습니다.
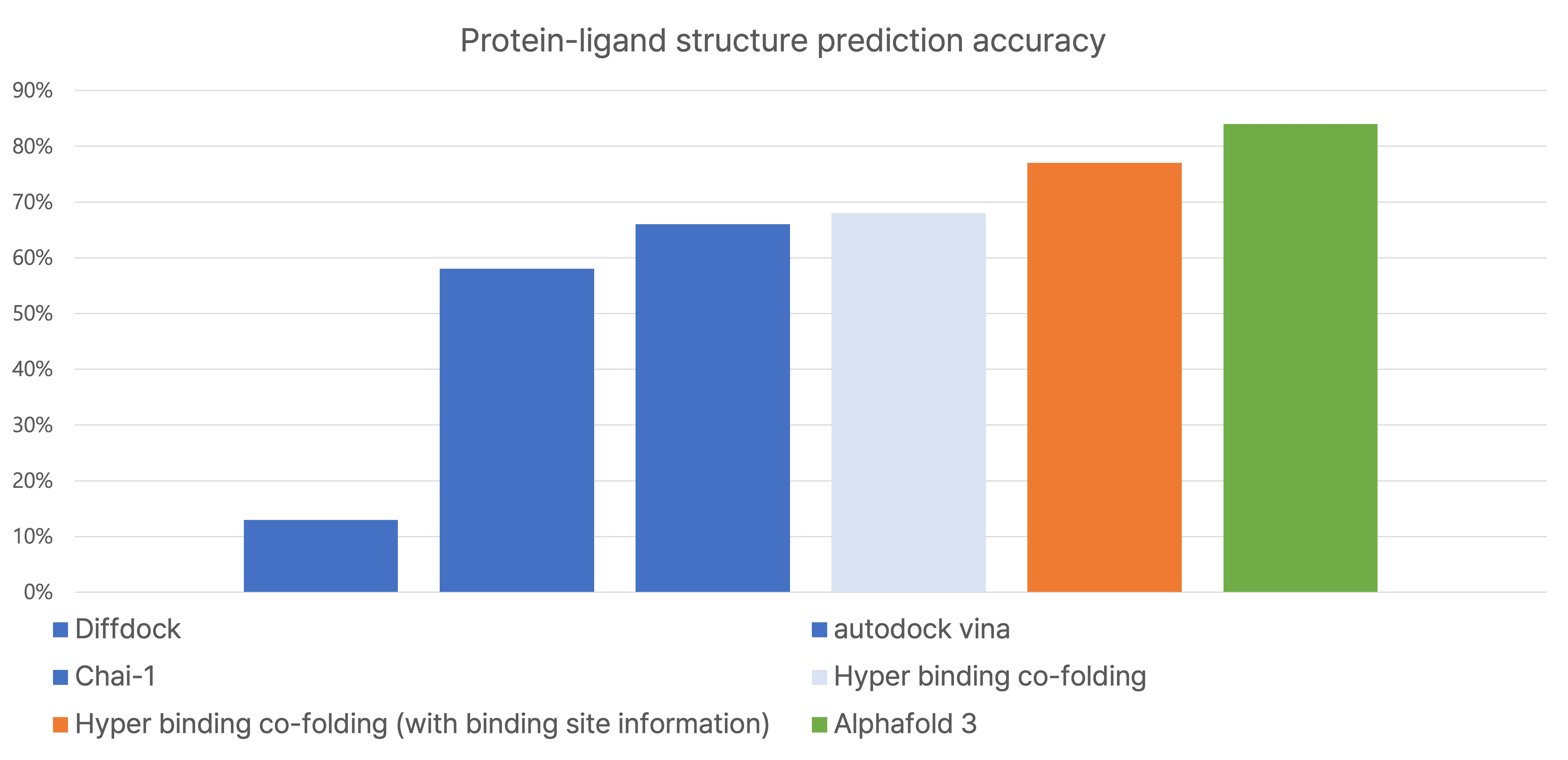
- 리간드-단백질 활성 예측 정확도: 단백질 서열 정보만으로도, 실험적으로 가장 잘 선택된 단백질 구조를 사용했을 때의 예측 정확도를 능가하는 결과를 보였습니다. 이는 실험 연구자들이 실제 연구 환경에서 체감하는 성능 향상에 크게 기여할 것으로 기대됩니다.
4. 사용자 편의성 비교
| 기능 | Hyper Binding Co-folding | AlphaFold3 | Chai Discovery |
|---|---|---|---|
| Induced fit 고려 | O | O | O |
| RNA 예측 | O | O | O |
| DNA 예측 | O | O | O |
| 단백질 키워드 검색 지원 | O | X | X |
| 다양한 결합 위치 정보 제공 | O | X | X |
| 전처리된 결합 부위 설정 제공 | O | X | X |
| 약물-단백질 결합 에너지 예측 | O | X | X |
혁신적인 Docking, Hyper Binding Co-folding 1주 무료 체험 가능
Hyper Binding Co-folding 기술은 구조 생물학, 화학정보학, 약물 디자인 경험이 부족하더라도, 신약 개발에 관심 있는 다양한 연구자들이 손쉽게 활용할 수 있도록 설계되었습니다. 특히 다음과 같은 분들에게 큰 도움이 될 수 있습니다.
- 단백질 3D 구조 없이 약물 결합 가능성을 예측하고 싶은 실험 연구자
- 신규 타겟에 대한 탐색이나 First-in-class 약물 개발을 시도하는 초기 단계 연구자
- Molecular Glue, PROTAC 등 복합 구조 기반의 최신 신약 전략을 연구하는 팀
- 전통적인 도킹 도구의 정확도나 복잡한 설정에 부담을 느끼셨던 분들
기존 도킹 기술의 한계를 넘어, Co-folding 기반의 접근법은 단백질 서열과 리간드 구조만으로도 높은 정확도의 예측이 가능하며, 복잡한 구조 준비나 결합 위치 지정 없이 바로 활용할 수 있습니다.
현재 Hyper Binding Co-folding 기술은 하이퍼랩(HyperLab) 플랫폼 내에서 제공되고 있으며, 누구나 1주일 동안 무료 체험해볼 수 있습니다.
신약 개발을 위한 구조 예측이 보다 직관적이고 쉬워지는 경험, 지금 바로 하이퍼랩을 통해 직접 확인해 보시기 바랍니다.
AI 신약개발 플랫폼 하이퍼랩