Hyper Screening X : World's Largest Molecular Library Powers the Future of Drug Development


Hyper Screening X: Virtual Screening of a 7-Trillion Compound Library with Deep Learning
Virtual screening is a widely used method in the early stages of drug discovery to identify promising hit compounds. With advancements in synthetic methods, the size of virtual screening libraries has grown significantly, reaching tens to hundreds of millions of compounds. Intuitively, the larger the library, the higher the chances of discovering novel and effective hits.
Recently, the emergence of ultra-large virtual libraries has pushed library sizes to an astronomical scale—trillions of compounds. In this article, we’ll explore how to efficiently navigate a virtual library of 7 trillion molecules.
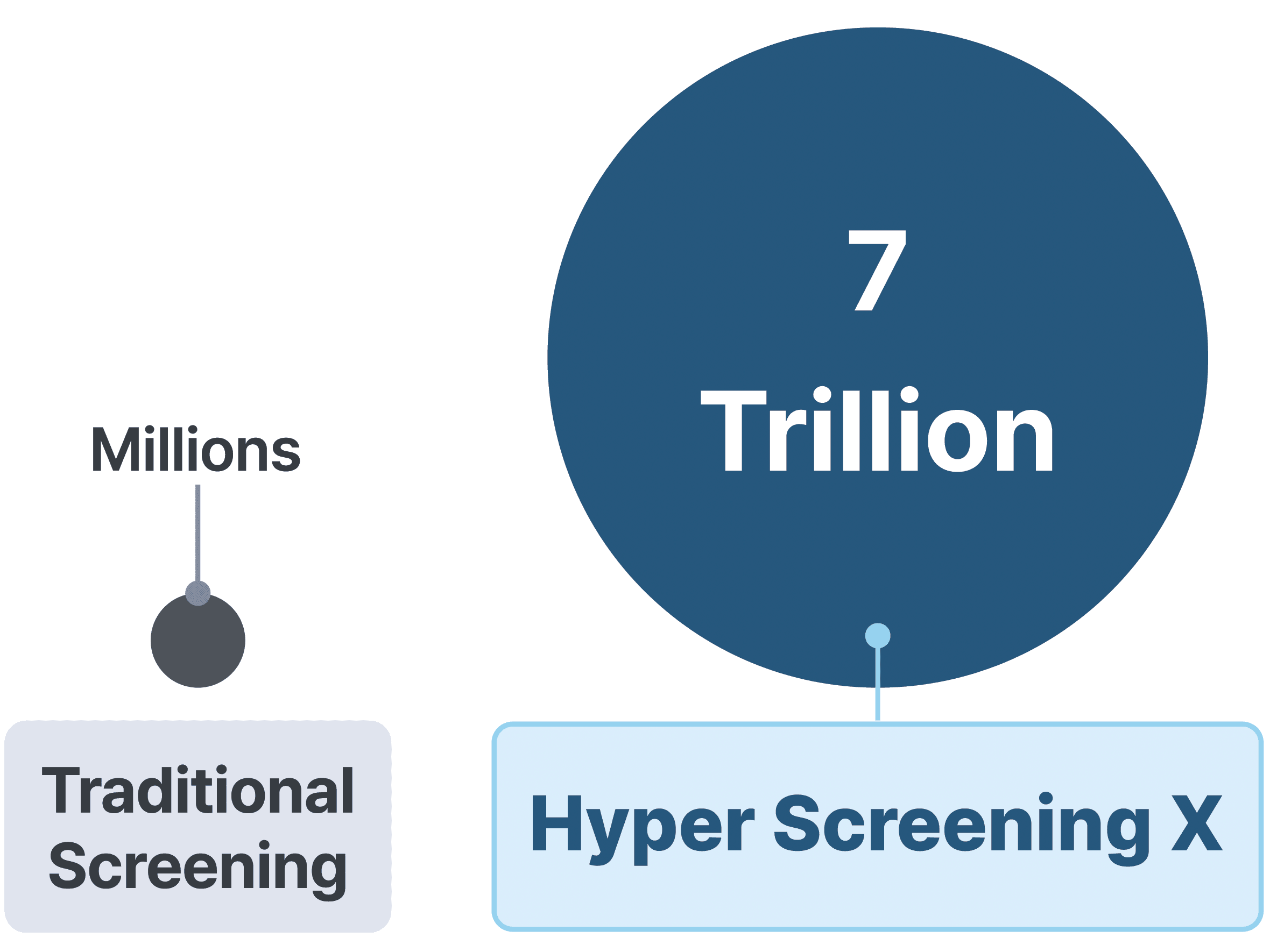
Why Deep Learning Is Essential
Let’s take structure-based virtual screening using docking as an example. If it takes 10 seconds to evaluate one molecule, screening 1 million molecules would require 10 million seconds—roughly 115 days. That’s over three months! To speed things up, parallel computing with multiple machines is often employed. With 100 computers, the time could theoretically be slashed to 1/100th, or about 1.15 days. That’s a manageable wait. For 10 million compounds, it’d take 11.5 days—still reasonable. Traditional methods can handle tens of millions of compounds this way.
But beyond that, it gets tricky. Screening 10 billion molecules with 1,000 computers would take 3 years. For 7 trillion? That’s 2,000 years! No one’s waiting that long for virtual screening results. Clearly, we need a smarter, more efficient approach to tackle a 7-trillion-compound library. Enter RxnFlow.
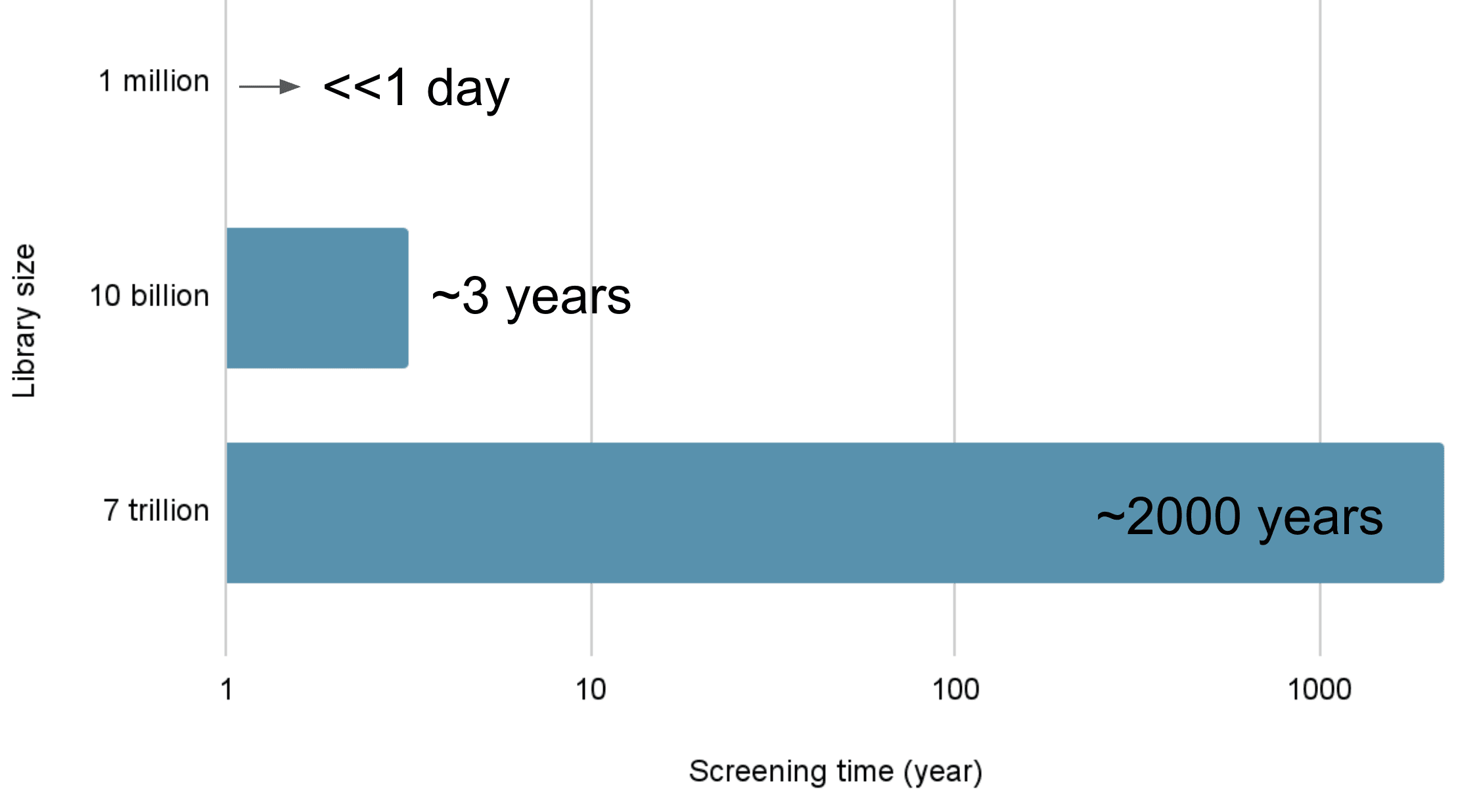
RxnFlow: Overcoming Time with Deep Learning
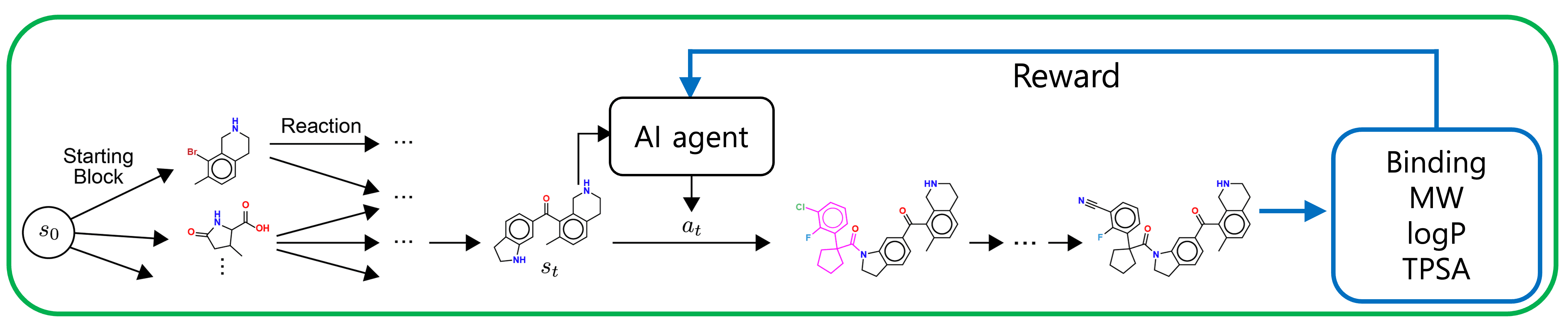
Let’s start with how virtual libraries work. These libraries are generated from combinations of chemical reactants and reactions. If we could predict which reactant-reaction pairs are likely to yield high target scores without evaluating every single one of the 7 trillion molecules, we’d be able to pinpoint the best combinations efficiently.
This is where deep learning shines. By analyzing data, it can uncover relationships between reactant-reaction combinations and target scores. Some building blocks likely contribute more to boosting scores than others. This insight tells us it’s smarter to focus exploration on high-potential combinations rather than treating all possibilities equally.
For this task, GFlowNet stands out. While reinforcement learning (RL) was once the go-to, it’s been criticized for getting stuck exploring only around specific molecular structures, missing broader diversity. GFlowNet, on the other hand, excels at identifying diverse modes (in this case, varied molecular structures), outperforming RL and gaining significant attention.
- What is GFlowNet?
- GFlowNet (Generative Flow Network) combines the strengths of reinforcement learning and generative models, aiming to efficiently sample from distributions with multiple modes.
- Unlike RL, which fixates on narrow regions, GFlowNet explores a wide chemical space, generating diverse molecular structures. It quickly learns to produce candidates that meet specific goals (e.g., high-activity molecules), accelerating the discovery of drug candidates and streamlining the drug development process.
Results Powered by Deep Learning
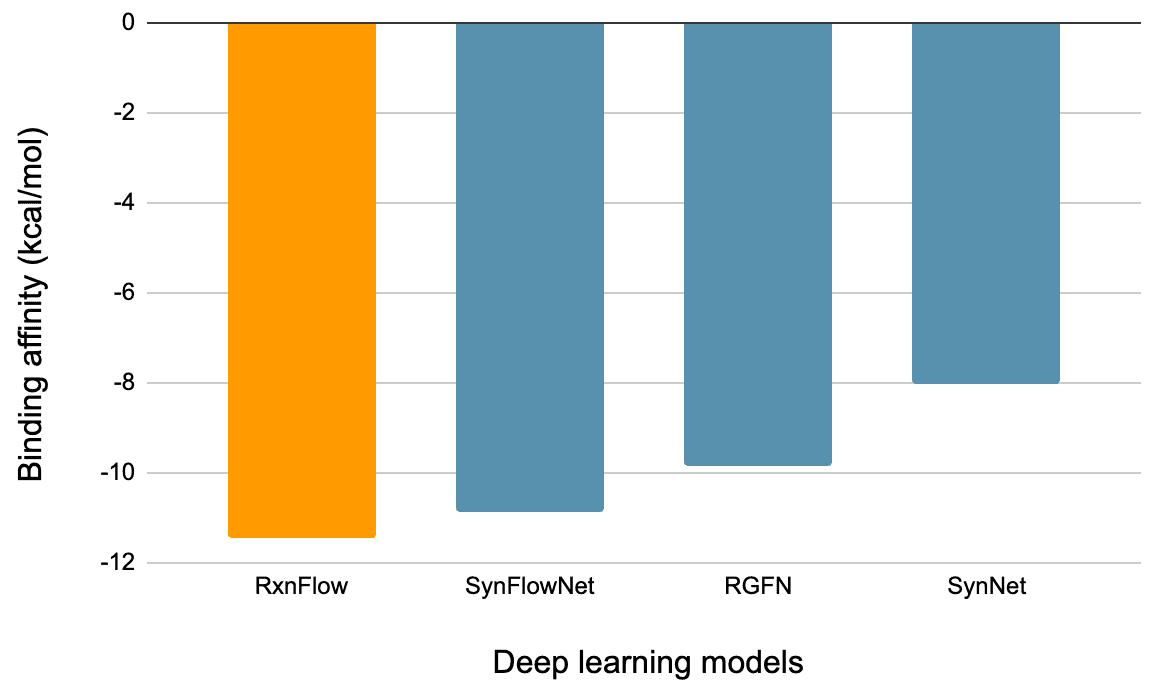
The first performance metric is binding energy—the lower, the better, as it indicates the AI has identified superior compounds. As shown in the table below, molecules generated by RxnFlow exhibit lower binding energies compared to those from other deep learning models.
Next up is synthetic feasibility. The table below shows the percentage of designed molecules that are synthesizable, assessed using AiZynthFinder (Journal of Cheminformatics, Volume 12, Article 70, 2020), a synthesis route prediction tool. A higher percentage means the model excels at designing practical molecules. RxnFlow achieves over 60%—a standout result, especially given the criticism that many AI-generated molecules lack synthetic feasibility.

This figure illustrates an example of a molecule and its synthesis route identified by RxnFlow. Researchers using Hyper Screening X can either synthesize the molecule themselves based on this route or outsource it to eMolecules.
Hyper Screening X leverages the exceptional RxnFlow as its core technology. For rapidly identifying hits in early drug discovery, exploring a 7-trillion-compound library with Hyper Screening X is the optimal solution.
When Will Hyper Screening X with Deep Learning Be Available?
HyperLab is currently preparing the launch of Screening X, which you’ll be able to experience soon. Sign up now to access binding, screening, design, and ADME/T tools to support your research. We’ll notify you by email when Hyper Screening X is released—start for free today!
AI-Powered Drug Discovery Platform: HyperLab
- [Free Trial] https://buly.kr/6MrAGFP
- [Contact Us for Implementation] https://abit.ly/6tr1uz