The 30-Year-Old Secret Behind AlphaFold's Success


You’re more powerful than you think
Sometimes it takes a while for a person to be recognized for their true worth. Steve Jobs, who has been called the most revolutionary entrepreneur in human history, went through the ordeal of being kicked out of Apple, the company he co-founded. He was able to bounce back after the company was in crisis and eventually prove his worth with a string of innovative products, including the iMac, iPod, iPhone, and iPad. Like Jobs' life, it shouldn't be surprising that behind some people's success stories lies a turbulent life.

AlphaFold, the winner of the 2024 Nobel Prize in Chemistry, has a story behind it. An innovative deep learning technology that predicts the three-dimensional structure of proteins, AlphaFold uses a technique called Multiple Sequence Alignment (MSA) to improve its accuracy dramatically. While MSA may not be a household name, it is a traditional technique used in the bioinformatics community to predict the function of proteins. The ancestor of MSA, ClustalW, was developed in 1994, so it's safe to say that the foundation of MSA was laid over 30 years ago.
Before the spotlight shone on AlphaFold's success, MSA was not only unknown to the general public but also slowly falling out of favor in the bioinformatics community. So why has MSA become so important again? In this post, we'll take a look back at what MSA was and is and discuss how a 30-year-old technology could play a big role in making AlphaFold's performance so dramatic.
What is Multiple Sequence Alignment (MSA)?
MSA is a method of aligning three or more biological sequences (DNA, RNA, proteins, etc.) to analyze their common ancestry, functional domains, and evolutionary relationships. In this post, we'll focus on proteins, which may sound complicated, but it's simply the process of finding commonalities in multiple protein sequences. In other words, MSA is a technique that aligns the amino acid sequences of various proteins at once, allowing you to see at a glance the similarities and differences between each sequence.

Proteins, the intermediaries that mediate life events, are made up of sequences of dozens to thousands of amino acids. These amino acid sequences can change during the evolution of life due to mutations in genes. But what if there are regions of the sequence that are conserved across species?
These conserved regions are likely to be associated with the protein's primary function, whereas regions with non-conserved sequences are unlikely to have a significant impact on maintaining the phenomenon of life. If a sequence that is essential to the maintenance of a life phenomenon were to change and become nonfunctional, the species would have been wiped out by natural selection. If a particular gene or protein sequence is consistently conserved across species, this suggests that it likely plays an important functional role. In addition to studying protein function, results from MSA can be used in a variety of biological studies, including analyzing the genetic causes of disease, evolutionary phylogenetic analysis, and more.
The Rise and Fall of MSA
An exhaustive history of MSA would take more than one post, so we'll just briefly mention some of the technologies that played a role in its evolution. While the concept of MSA has always existed, the first sign of a major turning point came in 1985.
1985: The FASTA format emerges
FASTA is a combination of the words “Fast” and Alignment. Since FASTA was originally a sequence alignment program, the name was chosen to emphasize that sequence alignments could be performed quickly. The file format used as input to FASTA is still widely used today as the FASTA format (fasta). The FASTA format is designed to be simple and efficient and consists of two elements.
- a header line: starts with > and contains the name or description of the sequence.
- sequence data: the protein (or nucleic acid) sequence, written as a sequence.
👨🏻💻Fasta file format for P0DK86 downloaded from UniProt
P0DKB6 RecName: Full=Mitochondrial pyruvate carrier 1-like protein; MARMAVLWRKMRDNFQSKEFREYVSSTHFWGPAFSWGLPLAAFKDMKASPEIISGRMTTA LILYSAIFMRFAYRVQPRNLLLMACHCTNVMAQSVQASRYLLYYYGGGGAEAKARDPPAT AAAATSPGSQPPKQAS
Since quickly sorting through many sequences is at the heart of MSA, it's not unreasonable to think of the FASTA format as the first key to MSA.
1990: Development of BLAST
BLAST (Basic Local Alignment Search Tool) is a sequence alignment tool that uses local alignment to find similarities between a given sequence and other sequences in a database. BLAST is much faster than traditional alignment algorithms because it only aligns the most similar parts of a sequence rather than the entire sequence. By detecting seeds based on short word units and expanding the seeds to find regions of high local similarity, the BLAST algorithm was able to focus more on the important parts rather than the entire sequence. BLAST is essentially a pairwise sequence alignment (PSA) tool that determines the similarity between two sequences, but it plays an important role in assisting the MSA algorithm that is developed later. It can be summarized in two main ways.
- Before performing MSA, quickly search and extract sequences similar to the MSA target sequence from a large protein database.
- Cluster sequences based on BLAST results to help the MSA algorithm work efficiently based on the evolutionary relatedness of the sequences.
1994: ClustalW sets the standard for MSA
ClustalW is a very popular program in bioinformatics. The “W” in ClustalW stands for weighted because ClustalW works by weighting sequences based on similarity between them. ClustalW uses a method called Progressive Alignment. It initially performed pairwise alignments and progressively generated multiple alignments based on them, as well as generated a guide tree representing evolutionary relationships based on the similarity between sequences and determined the order of alignment based on this guide tree. These concepts had a major impact on later MSA programs (e.g., Clustal Omega, MUSCLE, MAFFT, etc.) and are still widely used in today's MSA tools.
Although faster and more sophisticated tools are now widely used, ClustalW is considered a historical milestone that set the standard for MSA because it influenced subsequent algorithms. It is based on ClustalW that AlphaFold's secret weapon is described in the title as having been developed 30 years ago. ClustalW even had a great user interface, which ushered in a golden age of MSA-based research and made MSA an essential tool for studying the function of proteins.
The MSA crisis brought on by NGS and deep learning
Once thriving, MSA research faced major challenges in the 2000s and 2010s.
In the 2000s, advances in next-generation sequencing (NGS) technology began to generate massive amounts of sequence data. Traditional MSA tools proved insufficiently fast and memory-efficient to handle large amounts of sequence data, as the resources required for computation grew exponentially with the number of sequences. Of course, MSA is still used for bioinformatic precision analysis, such as protein domain analysis and exploration of conserved gene regions, but as the technology paradigm shifts to “big data,” new techniques have emerged to replace or complement MSA in large-scale sequence data analysis. Profile-based alignment, hidden Markov models (HMMs), NGS read mapping techniques, and data clustering-based techniques have received much attention.
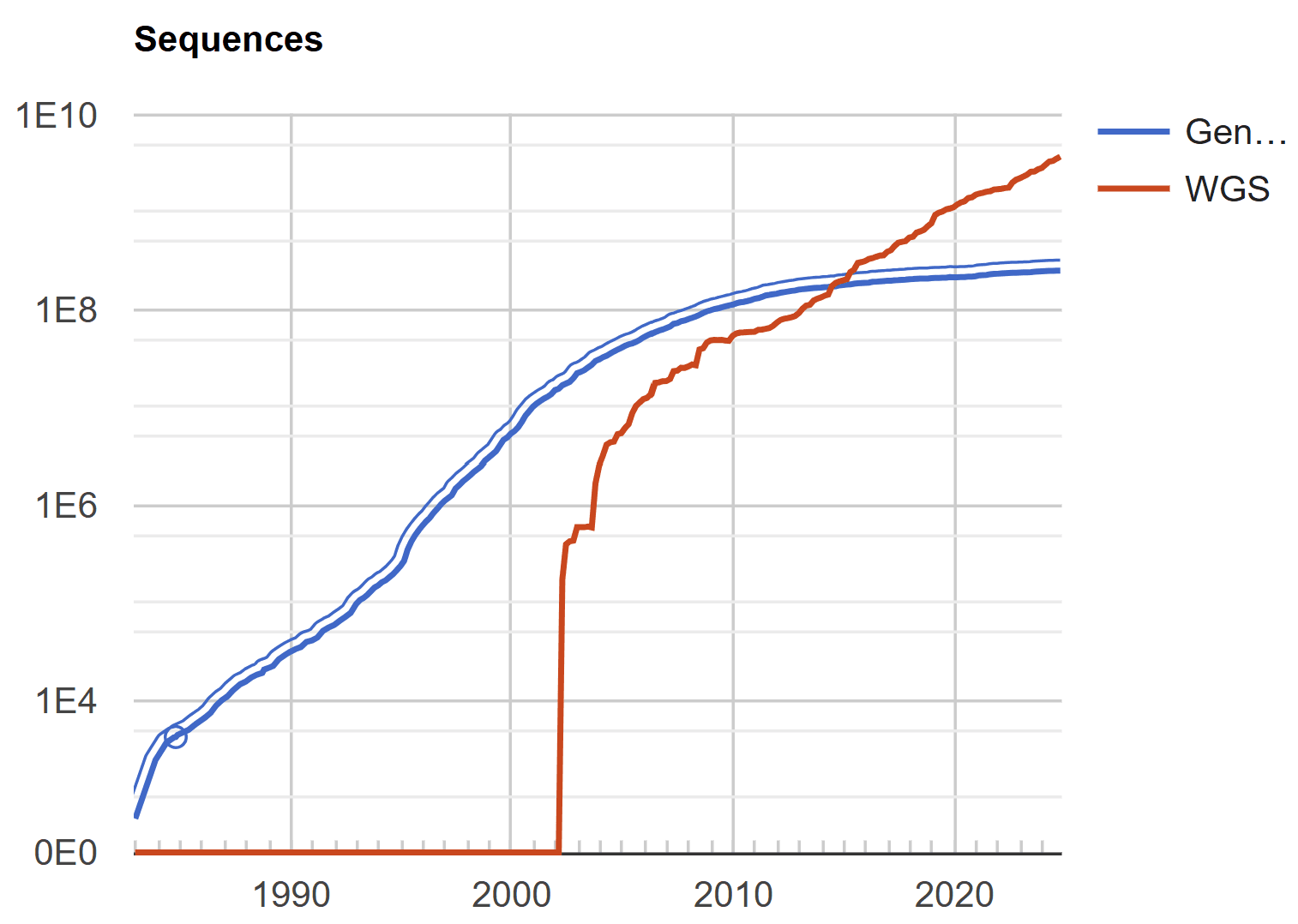
In the 2010s, MSA faced another challenge when deep learning emerged as a new technique for analyzing biological sequence data. Unlike MSA, which faced threats from the rise of NGS, deep learning perfectly complemented NGS's massive data production. As deep learning models learn from the massive sequence data generated by NGS, they can identify and predict patterns among sequences much more efficiently than traditional MSAs. These models have been very effective at discovering complex biological patterns through training on large amounts of data, and they are also more efficient than MSAs in terms of computational resources.
Models such as Recurrent Neural Networks (RNNs) and Convolutional Neural Networks (CNNs) were well suited to understanding and predicting sequence data. The growth of these deep learning-based sequence trait prediction models culminated in the late 2010s with the introduction of transformer-based models on sequence data. Transformer-based models outperformed not only MSA but also RNN- and CNN-based models.
MSA is back with a vengeance with AlphaFold
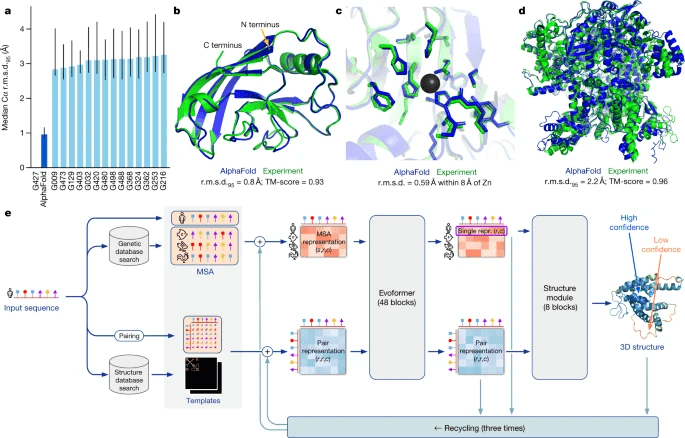
Did they say I knelt to gain momentum? Interestingly, the same deep learning that drove MSA to the brink has brought it back from the dead. AlphaFold, a leader in deep learning-based protein structure prediction models, uses an MSA model called MMSeqs2 to extract evolutionary information from protein sequences and use it as input to deep learning models. MSA plays two main roles in learning.
- Provide evolutionary information MSA can identify structurally and functionally important regions of a protein. Evolutionarily conserved regions play an important role in helping proteins maintain a stable three-dimensional structure. AlphaFold learns these conserved patterns through MSA.
- Co-evolutionary Signal Extraction Co-evolutionary interactions between multiple sequences obtained through MSA are used to predict contacts between amino acids in a protein. For example, if two amino acid residues exhibit evolutionarily interdependent variation, they are more likely to be located close together in three-dimensional space. AlphaFold learns these signals to model the folding structure of a protein.
With MSA as input, which identifies conserved regions between protein sequences and helps us understand the functional properties of proteins, AlphaFold outperformed existing protein structure prediction models, predicting the structures of tens of thousands of proteins whose structures have yet to be determined, and earning the 2024 Nobel Prize in Chemistry for its work. AlphaFold's success demonstrates the powerful synergies that can be created when traditional bioinformatics techniques like MSA are combined with the latest deep learning technologies. Read the more technical story of AlphaFold in this blog post.
Until the spotlight comes on
As with any revolutionary technology, it's often the case that it's not well understood at the time of its inception, and only later is its true value discovered. Even CRISPR-Cas9-based gene editing, which has revolutionized the world of genetics, was just an interesting defense mechanism used by bacteria to fight off viruses when it was first discovered in 1987. AI, the paradigm of today's technology, was also a minor discipline before advances in computer hardware, including GPUs, and the discovery of backpropagation algorithms, which eventually unlocked its true value. I think MSA has also been rediscovered today because its intrinsic value is greater than people thought 30 years ago.
We, the HITS Hyperlab team, will continue to develop better technologies and prepare more valuable services.