단백질 구조 기반의 약물 설계 - 배경 지식 편


약물 설계는 논리적이고 이론적인 지식을 활용해 새로운 약물의 구조를 설계하는 과정을 말합니다. 과거에는 주로 실험을 통해 얻은 데이터에 의존했지만, 최근에는 AI 기술의 발전으로 시뮬레이션과 예측을 통해 효율성과 성공 가능성을 높이고 있습니다.
약물 설계는 활성, 선택성, 신규성 등 신약 개발을 위한 다양한 지표를 개선하는 데 활용됩니다. 일반적으로 약물은 표적 단백질에 결합하여 그 활성을 조절함으로써 약효를 발휘하기 때문에, 표적 단백질의 결합 부위(binding site)에 안정적으로 결합할 수 있는 분자 구조를 탐색하는 과정은 약물 설계에서 핵심적인 단계라고 할 수 있습니다.
그렇다면 이러한 분자 구조는 어떻게 찾을 수 있을까요? 이론적으로 존재 가능한 저분자 유기 화합물의 수는 약 10⁶⁸개에 달한다고 알려져 있습니다. 바닷가와 사막의 모래알 개수가 약 7×10²⁷개라는 점을 고려하면, 아무런 정보 없이 우연히 약물을 찾을 확률은 모래사장에서 바늘을 찾는 것보다도 훨씬 낮다는 사실을 실감할 수 있습니다.
게다가 화합물이 실제 약물이 되기 위해서는 ADME/T 등 다양한 조건들도 충족해야 합니다. 그렇기 때문에 어떻게 하면 효율적으로 약물을 설계할 수 있을지에 대한 고민은 피할 수 없는 과제가 됩니다. 약물 설계를 포함한 모든 연구에서 정보는 풍부할수록, 그리고 이를 얼마나 효율적으로 활용하느냐에 따라 더 나은 성과를 기대할 수 있습니다. 이번 글에서는 AI의 예측과 분석을 활용한 효율적인 약물 설계 방법인 CADD(Computer-Aided Drug Design)에 대해 살펴보겠습니다.
단백질-화합물, 무엇이 결합 에너지를 결정하는가?
화합물이 표적 단백질에 안정적으로 결합하기 위해서는 물이라는 생물학적 환경, 단백질의 유연한 움직임 등 다양한 조건들을 고려해야 합니다. 그러나 가장 기본적으로 중요한 요소는 결합 부위에 잘 들어맞는 형태(shape)와 전자 분포(charge distribution)를 갖추는 것입니다. 이러한 요소들이 중요한 이유는, 결국 단백질과의 안정적인 상호작용을 형성해야만 약물로서의 기능을 제대로 수행할 수 있기 때문입니다.
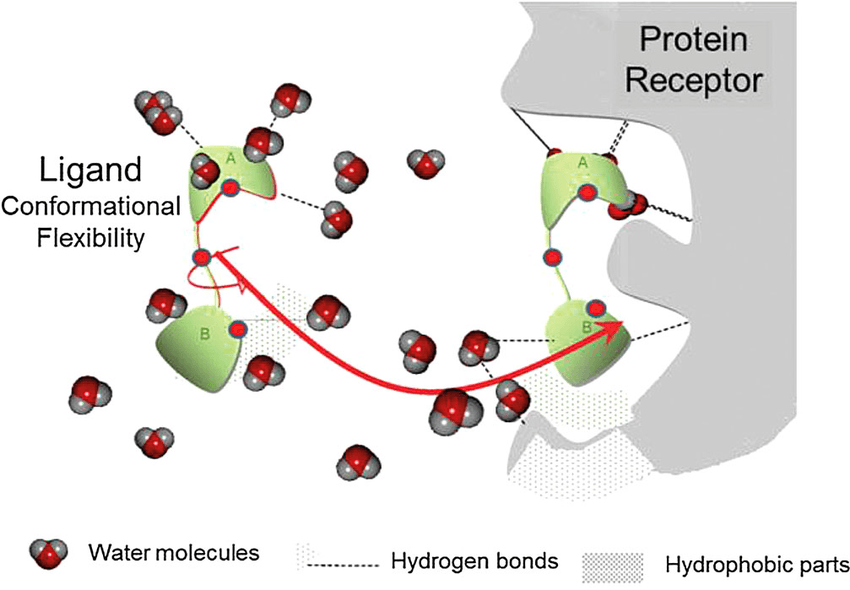
1. 형태
우선 형태(shape)에 대해 생각해봅시다. 분자의 형태가 표적 단백질의 결합 부위에 잘 맞을 경우, 단백질과의 접촉 면적이 극대화되어 더 많은 상호작용을 형성할 수 있는 기회를 얻게 됩니다. 또한, 결합 부위에 꼭 맞는 형태는 결합된 분자의 움직임을 제한해, 보다 안정적인 상호작용을 이루는 데 유리합니다. 특히, 소수성 상호작용(hydrophobic interaction)과 같은 비특이적 상호작용은 이러한 접촉 면적과 결합 구조의 안정성에 큰 영향을 받습니다.
2. 전자의 분포
전자 분포는 단백질과의 상호작용을 결정짓는 요소로, 작용기(functional group)라고 이해하셔도 됩니다. 예를 들어, OH나 NH와 같은 작용기는 수소 결합(hydrogen bonding), COO⁻나 N⁺는 이온 결합(ionic interaction), benzene 고리는 π-π 상호작용 또는 소수성 상호작용(hydrophobic interaction)을 형성할 수 있습니다. 이러한 상호작용은 기본적으로 두 분자 또는 작용기 사이의 상호작용이기 때문에, 수소 결합의 경우처럼 donor와 acceptor가 짝을 이루어야만 발생합니다. 따라서 표적 단백질 결합 부위의 특정 아미노산 잔기(residue)와 안정적인 상호작용을 만들기 위해서는, 화합물 내에 해당 상호작용을 형성할 수 있는 작용기가 적절한 위치에 있어야 합니다.
단백질-화합물 결합 구조 예측
단백질과 화합물의 3차원 결합 구조는 화합물의 형태나 전자 분포를 분석하는 데 가장 중요한 정보입니다. 이러한 결합 구조를 통해 단백질과 화합물 사이의 상호작용을 정밀하게 분석할 수 있고, 이를 바탕으로 목적에 맞는 약물 설계를 진행할 수 있습니다. 예를 들어, 새로운 구조를 도입하거나 기존 구조를 변형해야 할 위치를 파악하고, 적절한 작용기를 선택할 수 있습니다. 하지만 실제로 실험적 방법(x-ray 결정학 등)을 통해 결합 구조가 밝혀진 경우는 매우 드뭅니다. 따라서 일반적으로는 분자 도킹(molecular docking) 기법을 활용해 단백질과 화합물 간의 결합 구조를 예측하게 됩니다.
Docking은 입력 정보 준비, 결합 구조 및 에너지 예측 (binding energy), 결과 분석 3단계로 정리할 수 있습니다.
1. 입력 정보 준비
도킹(Docking)은 일반적으로 결합 부위(binding site)와 결합할 분자의 구조, 이 두 가지를 입력 정보로 사용합니다. 입력 파일의 형식은 사용하는 프로그램에 따라 다를 수 있으며, 특히 결합 부위의 정의는 도킹 알고리즘과 함께 결과의 정확도를 결정짓는 핵심 요소 중 하나입니다.
단백질은 정적인 구조가 아니라 주변 환경에 따라 side chain이나 backbone이 움직일 수 있는 유동적인 성질을 가지고 있습니다. 예를 들어, 키나아제(kinase)의 경우 저해제(inhibitor)의 타입에 따라 결합 부위의 형태가 크게 달라질 수 있습니다. 그러나 우리가 활용하는 X-ray 구조는 단백질이 가질 수 있는 다양한 구조 중 하나의 snapshot에 불과합니다. 따라서 예측하고자 하는 분자의 특성을 고려해 도킹에 사용할 단백질 구조를 신중히 선택하는 것이 중요합니다.
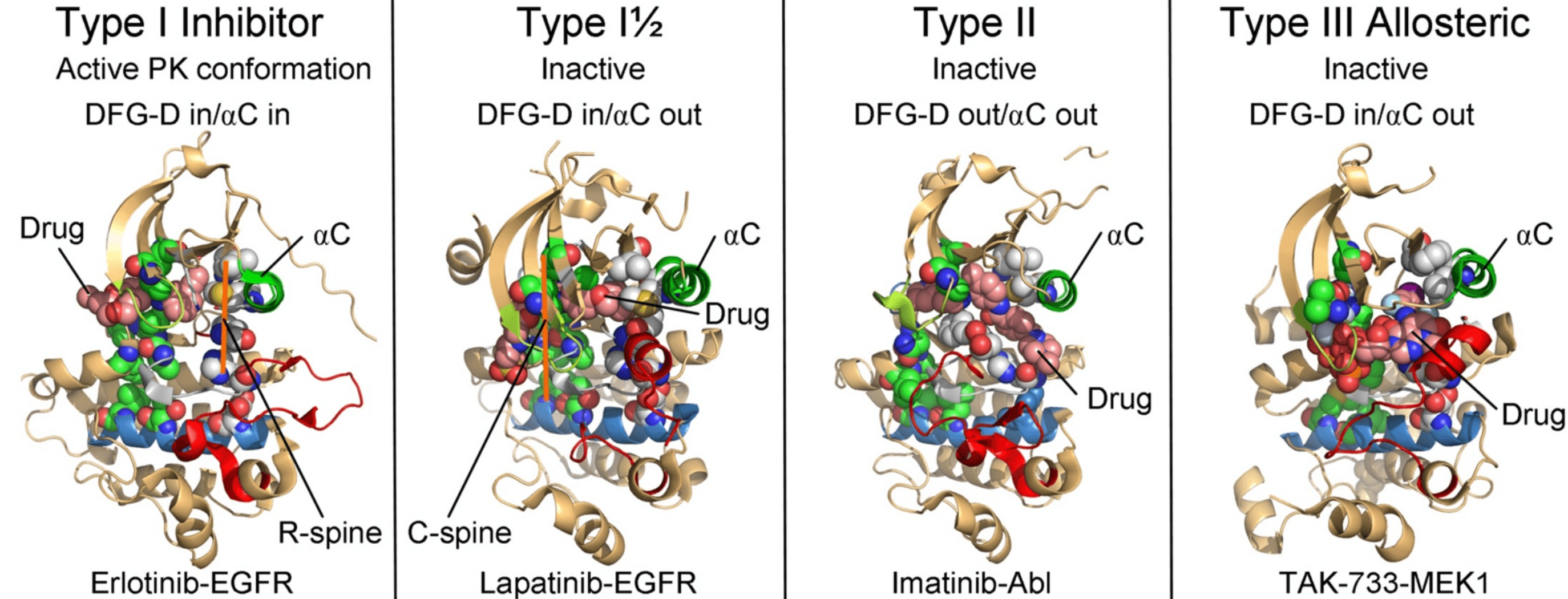
-
- 결합 부위는 기본적으로 하나 이상의 단백질로 구성되며 작용 기전에 따라 Heme과 같은 coenzyme과 Zn과 같은 금속을 포함 할 수 있습니다. 이들은 리간드와 상호작용하여 결합 구조와 안정성에 영향을 줄 수 있기 때문에 binding site 환경을 정의할 때 주의해야 합니다.
- 사용 목적에 따라 결합 부위의 크기와 위치를 조절할 수 있습니다. 결합 부위를 너무 넓게 잡으면 결합 구조를 탐색하는데 너무 긴 시간이 걸리거나 충분히 탐색하기 어렵고, 너무 작다면 분자 구조가 들어가 공간이 부족해서 결합 구조 예측이 실패할 수 있습니다. 예측하고자 하는 분자들의 크기를 고려하여 적절한 크기와 위치의 결합 부위를 정의하는 것이 바람직합니다.
- 예측하고자 하는 분자 구조는 전처리를 통해 입력 정보로 변환됩니다. 전처리는 salt 등 불필요한 정보 제거, pKa를 반영한 protonation과 deprotonation, rotatable bond 정의 등을 포함합니다.
2. 결합 구조 및 에너지 예측
- 입력 정보를 잘 준비했다면 예측은 알고리즘이 자동으로 처리해 줍니다.
3. 결과 분석
- 계산이 잘 마무리 되었다면 docking 결과를 얻을 수 있습니다. Docking 결과에는 기본적으로 분자의 결합 부위에 대한 결합 구조와 점수 (score)를 포함합니다. 결합 구조는 docking 프로그램에서 제공하는 도구를 사용하거나 무료로 제공되는 PyMOL 등을 사용하여 확인할 수 있습니다.
- Docking 프로그램이나 설정에 따라 다를 수 있지만 한 분자 당 10개 정도의 예측 된 결합 구조를 제공합니다.
- 점수는 결합 에너지(kcal/mol)를 의미하며, 음수 값을 가지며 절대값이 클수록 더 안정적인 결합을 이룬다고 해석할 수 있습니다. 즉, -4 kcal/mol보다는 -6 kcal/mol이 더 안정적인 결합을 나타냅니다.
- 주의해야 할 점은, 이 결합 에너지가 반드시 약물의 활성(IC50, Kd 등)과 동일한 의미는 아니라는 점입니다. 이는 약물의 작용 기전, 모델의 학습 목적, 그리고 성능과 관련이 있습니다.
- 약물이 competitive inhibitor라면 결합 에너지와 활성은 높은 상관관계를 가질 수 있습니다. 그러나 allosteric inhibitor처럼 결합이 단지 활성을 발휘하기 위한 하나의 조건일 경우, 결합 에너지 값만으로는 약물의 활성을 설명할 수 없습니다.
- Docking 알고리즘에서 점수를 계산하는 모델은 활성 예측보다는, 다양한 결합 구조를 탐색하는 과정에서 생성된 구조들 간의 비교 우위를 평가하는 데 더 큰 목적이 있습니다. 결합 구조의 안정성을 평가하는 점에서 활성을 일정 부분 설명해줄 수는 있지만, 이 점수를 어느 정도까지 신뢰할 수 있는지는 별도의 평가가 필요합니다.
- Docking 모델에 따라 결합 구조 예측과 활성 예측의 성능은 다를 수 있습니다. 특정 단백질에 대한 성능을 비교한다면 이는 더 큰 차이가 있을 수 있습니다.
- 따라서 약물 설계 과정에서 설계된 분자 구조의 결합 구조와 활성을 docking으로 평가하고자 한다면 사용하는 docking 방법이 얼마나 결합 구조와 활성을 잘 예측해 주는지 평가하는 과정이 필요합니다. 이는 단순히 docking 방법을 평가하는 목적에서 더 나아가 docking 환경을 최적화해 더 나은 결과를 얻기 위한 과정입니다.
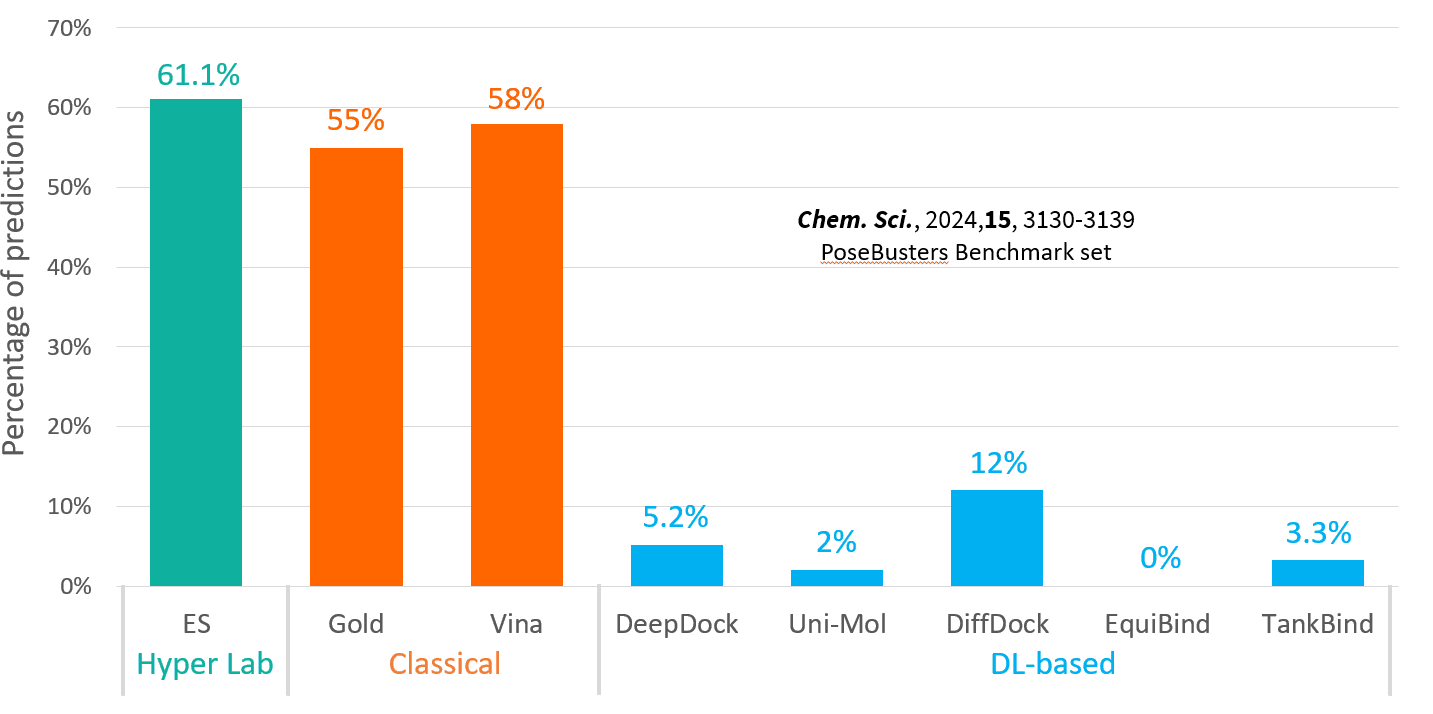
이제 단백질-약물 결합 구조를 예측할 준비가 되었습니다. 하지만 예측 정확성을 높이고 더 나은 결과를 얻기 위해서는 docking 과정을 좀 더 자세히 들여다 볼 필요가 있습니다. 다음 글에서는 docking 환경 설정의 최적화를 위한 전략들을 살펴 보겠습니다.