가상탐색을 위한 단백질 구조 선정 방법


이번 글에서는 self-docking과 cross-docking을 활용해 가상탐색을 위한 표적 단백질의 구조 (conformation) 선택 과정을 다뤄 보겠습니다. 이전 글에서는 구조 기반 신약 개발 연구(SBDD) 과정에서 binding site를 정의할 때 고려하면 좋을 사항을 소개했습니다. 아직 읽어보지 않으셨다면 참고해 보셔도 좋습니다.
가상 탐색
가상탐색은 수십만에서 수십억 개의 디지털 화합물 라이브러리에서 다양한 조건을 만족하는 소수(보통 수백 개)의 화합물을 선택합니다. 이후 이들 후보 물질을 실험으로 검증해서, 신약 개발 가능성이 있는 유효 물질을 찾는 것이 궁극적인 목표입니다.
(화합물 라이브러리의 선택 또한 매우 중요한 항목이지만, 이는 다른 글에서 다루겠습니다.)
화합물을 선별하기 위한 조건들은 연구 목적에 따라 다양하게 설정될 수 있습니다. 일반적으로 빠르게 계산할 수 있는 Lipinski’s rule 등의 항목을 우선적으로 적용해 초기 후보군을 빠르게 걸러냅니다. 물론 뒤로 갈수록 계산 비용은 높아지지만, 신뢰도가 높은 최종 후보 물질을 선별하게 됩니다.

단백질 약물 결합 구조 방식
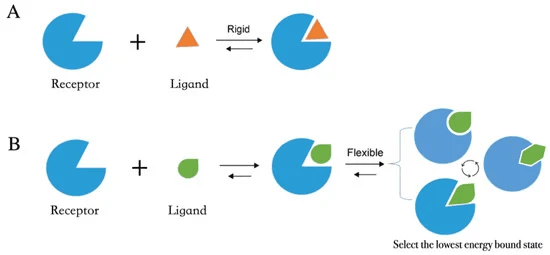
단백질 - 약물을 결합하는 방식에는 두 가지 모델이 있습니다.
단백질 약물 결합 구조 방식 1. Lock and key
우선 “Lock and Key” 모델이 있습니다. 이 모델은 단백질이 약물을 받아들일 수 있게끔, 이미 결정된 binding site 구조를 가지고 있다고 가정한 채 진행됩니다. 마치 자물쇠에 꼭 맞는 열쇠가 있어서, 자물쇠를 열거나 잠글 수 있는 것과 유사합니다. 상단의 그림을 참고하시면 더욱 이해가 쉬울 것입니다.
단백질 약물 결합 구조 방식 2. Induced fit
이후에는 단백질의 유동성이라는 중요한 특성을 반영한 “Induce Fit” 모델이 나왔습니다. Induce Fit 모델에 따르면 단백질과 약물은 상호작용을 통해 서로의 구조적 변화를 유도하고, 에너지적으로 안정된 결합 상태를 형성하게 됩니다.
더 나아가, 단백질이 본래 여러 가지 구조(conformations)를 취할 수 있다는 점에 주목한 Conformational Selection and Population Shift 모델도 등장했습니다. 이 모델은 리간드가 단백질의 다양한 구조 중 가장 잘 맞는 conformation을 선택하고, 이후 해당 구조가 안정화되어 우세해지는 과정을 설명하며, 현재 널리 받아들여지고 있습니다.
다행히도 우리는 ‘단백질과 약물이 어떻게 안정적인 결합 구조를 이루는가?’에 대해 정확하게 알 필요가 없습니다. 우리가 집중해야 할 질문은 “그래서 어떤 단백질 구조를 써야 하지?” 입니다. 이제 다시 구조 기반의 가상탐색으로 돌아가 보겠습니다.
도킹(Docking)
“그렇다면 어떤 단백질 구조를 써야 하지?”와 같은 질문은 도킹(docking)을 수행하기 위한 시작점이자 핵심이 되는 과정입니다. Docking은 단백질과 약물의 결합 구조를 예측하는 동시에, 구조 기반 신약 개발 연구(SBDD)의 출발점이 됩니다.
도킹(Docking)이란?
신약 개발에서 도킹은 분자 수준에서의 상호 작용을 분석하고, 이해하는 데 사용되는 컴퓨터 기반의 기술입니다. 보통 분자 구조를 모델링한 뒤, 특정 분자와 다른 분자 간의 상호 작용이 어떻게 이루어지는지 예측하고 이해하는 데 활용됩니다. 이를 통해 신약 후보 물질의 결합 매커니즘을 파악하고, 보다 효과적인 약물 설계 및 최적화에 활용할 수 있습니다.
도킹(Docking)을 위해 한 개 이상의 단백질 구조를 선택할 때, 올바른 의사 결정을 내리기 위해 참고해야 할 다양한 정보들이 존재합니다. 이번 글에서는 가장 이상적인 실험 조건에서 출발해, 실제 가상 탐색 환경에서 고려해야 할 변수들을 단계적으로 추가해나가는 방식으로 이를 살펴보겠습니다. 이러한 접근 방식은 도킹에 적합한 단백질 구조를 선택하는데 도움이 될 것입니다.
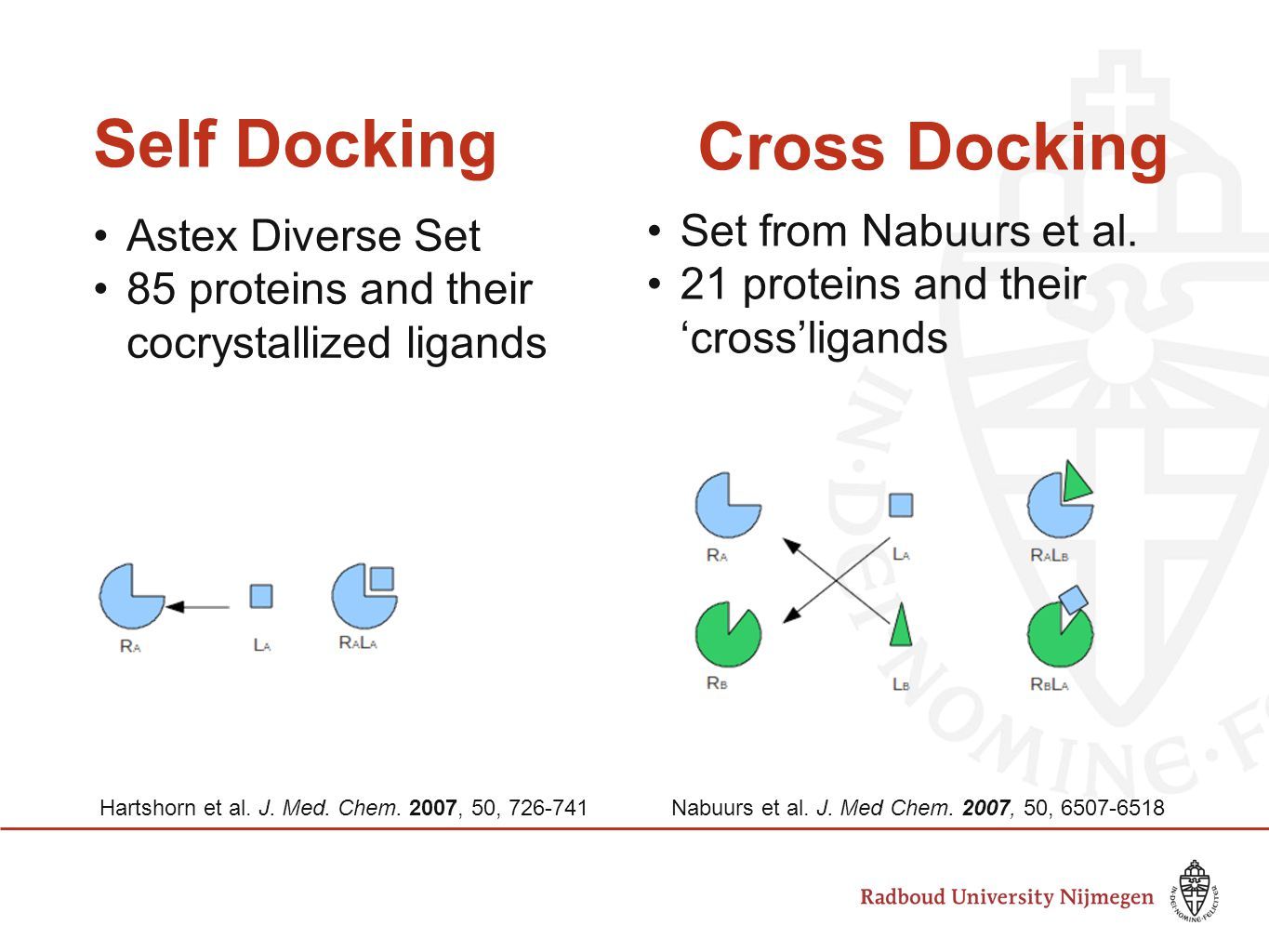
셀프 도킹(Self-Docking)
셀프 도킹(Self-docking)은 단백질-약물 x-ray 결합 구조에서 약물을 떼어낸 후, 도킹으로 x-ray 구조를 재현할 수 있는지 확인하는 과정입니다.
이 과정을 가장 먼저 수행하는 이유는, 가장 x-ray 결합 구조가 실험적으로 결정된 가장 이상적인 조건이기 때문입니다. 구조 자체의 오류가 적고, 도킹 알고리즘의 성능을 점검하기에 적합한 기준이 됩니다.
만약 셀프 도킹을 통해 x-ray 구조를 제대로 재현하지 못한다면, 사용한 여러 항목을 재검토해야 합니다. 일반적으로 약물의 실험과 도킹 구조의 RMSD가 2Å 이하일 경우, 결합 구조를 잘 예측한 것으로 평가합니다.
도킹 결과가 X-ray 결합 구조를 제대로 재현하지 못할 경우, 가장 먼저 단백질 구조, 특히 binding site의 상태를 점검해야 합니다. 실험구조라고 해서 항상 완벽한 것은 아니기에, 다음과 같은 요인들을 확인할 필요가 있습니다.
가장 먼저 alternative conformation입니다. 이전 글에서도 다뤘듯이, 단백질 구조에는 동일한 residue에 대해 두 가지 이상의 conformation이 존재할 수 있습니다. 이때, occupancy(존재 가능성)가 가장 높은 conformation이 일반적으로 선택됩니다. 대부분의 경우 문제가 없지만, occupancy 값이 비슷하거나 실제로 두 번째 conformation이 더 적절한 경우도 있으므로 주의 깊은 확인이 필요합니다.
또 다른 가능성은 x-ray 결과의 해상도(resolution) 문제입니다. X-ray는 단백질의 역동적인 움직임을 특정 시점에서 포착한 '스냅샷'에 해당됩니다. 해상도는 이 구조가 얼마나 정밀하게 측정되었는지를 나타냅니다. 일반적으로 resolution이 2Å 이하이면 큰 문제가 없지만, 그 이상이라면 아미노산 residue와 약물 사이가 너무 가까워 충돌하는 등의 문제가 있을 수 있습니다.
이런 경우 MD simulation을 통해 구조를 최적화하거나 다른 구조를 선택하는 등, 다양한 방식으로 대응할 수 있습니다. 한 PDB 파일에 동일한 단백질-약물 결합 구조가 두 개 이상 포함된 경우, 각 구조 간에 미세한 차이가 있을 수 있습니다. 이런 경우, 여러 구조를 비교 실험해 보는 것도 하나의 방법입니다.
구조적으로 문제가 없을 경우 도킹 알고리즘이나 옵션 설정을 검토해 볼 필요가 있습니다. 셀프 도킹으로 x-ray 구조가 재현되지 않는다면 binding site의 크기를 줄이거나 exhaustiveness (thoroughness)를 높이는 등 설정을 바꿔서 실험해 볼 수 있습니다.
하이퍼랩은 다양한 실험을 통해 최적화된 docking 환경을 제공하고 있습니다. 또한 정확성이 매우 개선된 자체 도킹(docking) 알고리즘인 ES 방법을 2024년, 올해 제공할 예정입니다.
크로스 도킹(Cross-Docking)
크로스 도킹(Cross-docking)은 단백질 A–약물 B 결합 구조에서 약물 B를 떼어낸 뒤, 이를 단백질 A–약물 C 결합 구조에서의 단백질 A에 도킹시켜 약물 B의 결합 구조를 재현할 수 있는지를 확인하는 과정입니다.
크로스 도킹은 셀프 도킹에 비해 단백질 구조라는 변수가 추가된 실험 환경입니다. 실제 가상 탐색에서는 결합 구조가 알려져 있지 않은 다양한 화합물을 사전에 선택한 단백질 구조에 도킹해야 합니다. 그래서 실제 상황에 보다 가까운 조건이라고 할 수 있습니다.
크로스 도킹의 목적은 바로 이 가상 탐색에 사용할 소수의 단백질 구조를 선택하는 것입니다. 단백질 구조는 1개 이상 선택할 수 있으며, 2개 이상을 선택해 다양한 구조를 반영하는 경우를 앙상블 도킹(Ensemble docking)이라고 합니다.

상단의 그림은 CK1 delta 23개 PDB 구조에 대한 크로스 도킹 결과 입니다.(10.3389/fmolb.2022.909499) X, Y 축은 각각 리간드와 단백질 구조를 나타냅니다. X 축 레이블은 PDB 코드 리간드 코드입니다. 오른쪽 컬러 바는 ligand의 x-ray 도킹 구조의 RMSD 값을 나타냅니다. 해당 수치가 0에 가까울수록, 즉 진한 파란색일수록 정확도가 높다고 볼 수 있습니다.
만약 단백질 구조 간 차이가 너무 커서 도킹만으로 리간드의 x-ray 결합 구조를 재현할 수 없다면, 대각선(self-docking)을 제외한 대부분이 붉은색으로 표시될 것입니다. 반대로 단백질 구조들이 매우 유사하여, 어떤 단백질을 사용해도 리간드 x-ray 구조가 잘 재현된다면 모두 파란색에 가까운 결과가 나타날 것입니다.
그런데 아래 그림에서는 파란색과 빨간색이 혼재되어 있습니다. 그렇다면 우리는 어떤 단백질 구조를 선택해야 할까요?
- 우선 파란색이 많이 나타나는, 즉 다양한 리간드의 결합 구조가 잘 재현되는 단백질 구조를 고릅니다. 저는 이러한 단백질을 '대표성이 있는 구조'라고 표현합니다.
- 하나의 단백질 구조만으로 대부분의 리간드 결합 구조를 정확히 예측할 수 있다면 이대로 마무리 해도 무방합니다. 하지만 특정 리간드에서 예측 정확도가 떨어진다면, 추가적인 단백질 구조를 고려할 필요가 있습니다.
- 이때는 가능한 적은 수의 단백질 구조로 최대한 많은 리간드의 결합 구조를 예측 할 수 있는 조합을 찾는 것이 바람직합니다. 무작정 많은 단백질을 사용하는 경우, 계산 시간은 물론 잘못된 결합 구조의 수도 폭발적으로 늘어날 우려가 있습니다. (탐색해야 할 화합물이 적어도 몇십만 개라는 걸 잊지 마세요!)
- 보통 도킹은 단백질-리간드 조합당 약 10개의 결합 구조를 제공합니다. 이 중 하나를 제외하고 정답이 없을 경우는 모두 잘못된 결합 구조입니다. 상단 그림에서는 4KBB-B와 6RCH를 선택하면 4HNF-16W를 제외하곤 모두 2Å 이내로 예측 가능함을 알 수 있습니다.
셀프 도킹과 크로스 도킹을 넘어 올바른 단백질 구조 선택까지
물론 위와 같은 방식으로 단백질 구조를 선택하더라도, 화합물 라이브러리에 있는 모든 화합물의 결합 구조를 정확히 예측할 수 있는 것은 아닙니다. 우리가 활용할 수 있는 정보는 한정되어 있고, 화합물 공간(Chemical space)은 이와는 비교 할 수 없이 방대하기 때문입니다.
이러한 단백질 구조 선택 과정은 현재 가지고 있는 정보를 최대한 활용하고, 보다 나은 결과를 얻기 위함입니다. 셀프 도킹과 크로스 도킹은 올바른 단백질 구조를 준비하기 위한 과정의 일부입니다. 다음 글에서는 핵심 단백질-약물 상호작용 분석과 활성 예측 과정을 다루도록 하겠습니다.