AlphaFold 3 이후, Docking 기술이 바꾸는 신약개발 패러다임


AlphaFold 3 이후, 왜 Co-folding인가
2021년, AlphaFold 2가 Nature에 공개되었습니다. 아미노산 서열만으로 단백질 3차원 구조를 원자 수준에 가깝게 예측해 구조생물학의 판을 바꿨고, CASP14에서도 기존 방법을 압도하는 성능을 보였습니다.
2024년 5월에 공개된 AlphaFold 3는 한 단계 더 나아가, 단백질뿐 아니라 DNA·RNA 같은 핵산, 소분자 리간드, 이온, 변형 잔기(PTMs)까지 포함한 복합체의 '공동(co-) 구조'를 확률적 확산(diffusion) 기반 아키텍처로 직접 예측할 수 있게 됐습니다. 즉, 리간드가 결합할 때 표적 단백질이 어떻게 미세하게 변형되는지까지 하나의 모델 안에서 동시에 다루기 시작한 것입니다.
이 변화는 전통적 ‘리간드를 고정된 단백질에 끼워 넣는’ 도킹 패러다임을, ‘상호작용하는 분자들을 함께 접는’ Co-folding 패러다임으로 이동시켰고, 초기 설계와 구조기반 최적화 단계 모두에 의미 있는 함의를 남깁니다.
이번 글에서는 이러한 배경 위에서 기존 도킹의 한계점을 짚고, 하이퍼랩의 하이퍼 바인딩 Co-folding이 실제 워크플로에서 장점과 적용 사례를 정리합니다. 간략한 내용은 아래 영상에서 보실 수 있습니다.
히츠의 ‘AlphaFold 3 이후, Docking 기술이 바꾸는 신약 개발 패러다임’ 웨비나를 토대로 작성되었습니다. 간략하게 편집한 영상도 있으니 참고해 주세요.
기존 도킹 방법의 한계점
1. 단백질 유연성 반영 어려움
대부분의 도킹 소프트웨어는 수천 개의 원자로 이루어진 단백질을 고정된(rigid) 구조로 가정합니다. 그러나 실제로는 리간드 결합 전후에 단백질이 수 Å(옹스트롬) 단위로 미세하게 변형되며, 이러한 작은 차이도 결합 포즈와 스코어에 큰 영향을 미칩니다. 단백질의 유연성을 정밀하게 다룰 수 있는 방법으로는 MD(분자동역학) 시뮬레이션이 있지만, 설정이 복잡하고 계산 속도가 수만 배 느려 실용성이 떨어집니다.
2. 바인딩 사이트 사전 지정 의존

계산량을 줄이기 위해 결합 위치(binding site)를 사전에 고정하는 것이 일반적인 관행입니다. 그러나 위치를 지정하지 않을 경우, 정확도가 절반 이하로 급격히 하락하는 문제가 자주 발생합니다(예: 약 50% → 20% 미만).
3. PDB 구조 선택 난제

하나의 타깃에 대해 PDB 구조가 여러 개 존재할 경우, 어떤 구조를 선택하느냐에 따라 예측 성능이 극단적으로 달라질 수 있습니다. 동일한 유도체 세트를 대상으로 하더라도, 구조에 따라 피어슨 상관계수가 0.86까지 나오기도 하고, 다른 구조를 사용하면 0.28까지 떨어지는 사례도 있습니다.
더 큰 문제는, 3D 구조 자체가 없거나 품질이 낮은 호모로지 모델만 있는 경우, 혹은 보고된 holo 구조가 최적 상태가 아닐 경우입니다. 이럴 때는 예측 정확도의 하락을 피하기 어렵습니다. 특히 새로운 기전을 겨냥한 first-in-class 과제일수록 이러한 문제가 더욱 두드러집니다.
기존 도킹의 한계를 극복한 하이퍼 바인딩 Co-folding
1. PDB 불필요, 서열 기반 결합 예측

하이퍼 바인딩 Co-folding은 기존 도킹처럼 고정된 단백질 구조에 리간드를 억지로 끼워 맞추는 방식이 아닙니다. 대신, 리간드 결합에 의해 유도되는 단백질의 미세한 구조 변화를 함께 예측합니다.
심지어 PDB 구조가 없어도 단백질 서열만으로 결합 포즈를 생성할 수 있어, 신약 개발의 초기 단계부터 유용하게 활용할 수 있습니다.
2. 결합 위치 자유도 확보
결합 위치를 미리 지정하지 않아도 유효한 포즈를 얻을 수 있습니다. 사전 지식이 있을 경우(예: 힌지 바인딩) 이를 반영하면 예측 정밀도를 더욱 높일 수 있습니다.
3. 구조 선택 자동화
전통적으로는 여러 PDB 중에서 가장 적절한 구조를 골라야 했지만, Co-folding은 동일 서열을 기반으로 구조를 직접 생성하기 때문에 이 과정 자체가 사라집니다. 더 나아가, 연구자가 신중히 선택한 최적의 holo 구조보다 Co-folding이 생성한 서열 기반 구조가 실제 활성 예측에서 더 우수했던 사례도 보고되었습니다.
4. 사용자 친화적 워크플로우
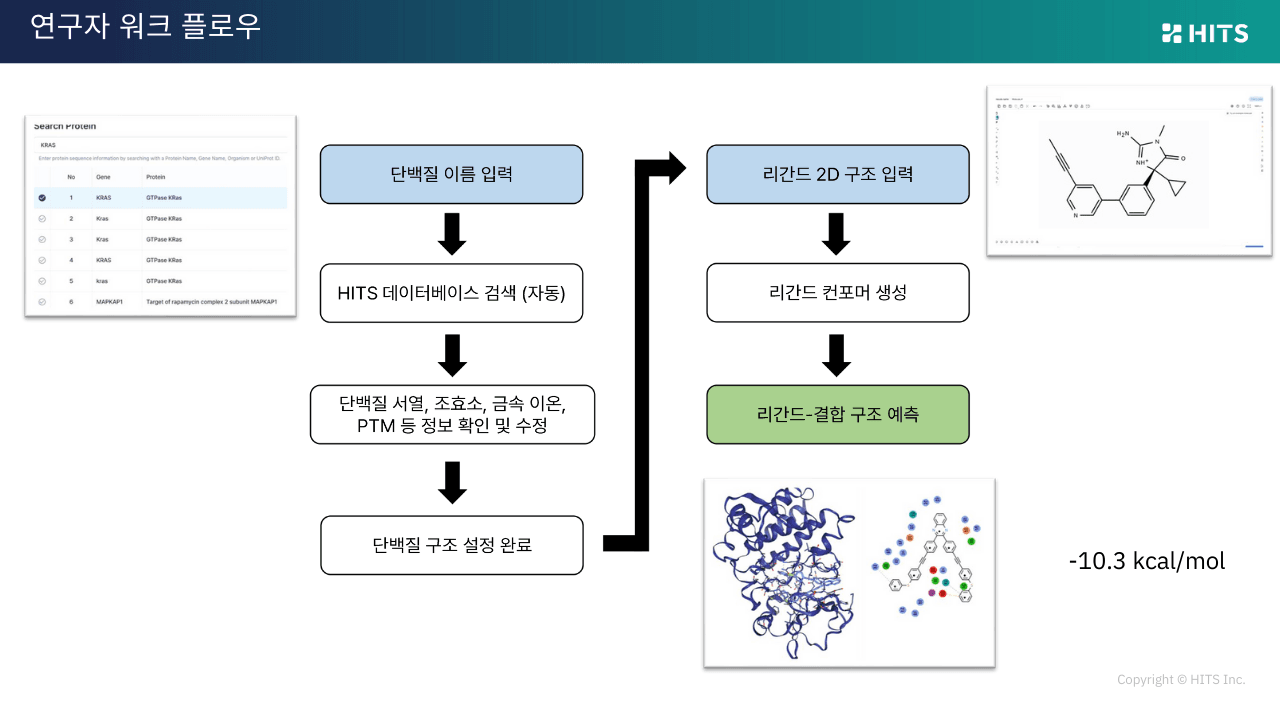
하이퍼랩에서는 단백질 이름(예: EGFR, KRAS, BTK)만 입력하면 유니프로트, 코팩터, 이온, PTM까지 자동으로 제안됩니다.
리간드는 직접 그리거나 파일을 업로드해 입력할 수 있으며, 결과는 3D 포즈, 2D 상호작용, 예측 에너지 형태로 즉시 확인할 수 있습니다. 실험 연구자도 별도의 학습 곡선 없이 바로 활용할 수 있도록 설계되어, 업무 효율성을 크게 높여줍니다.
결과 및 응용 사례
케이스 1. JAK2 계열 유도체 세트
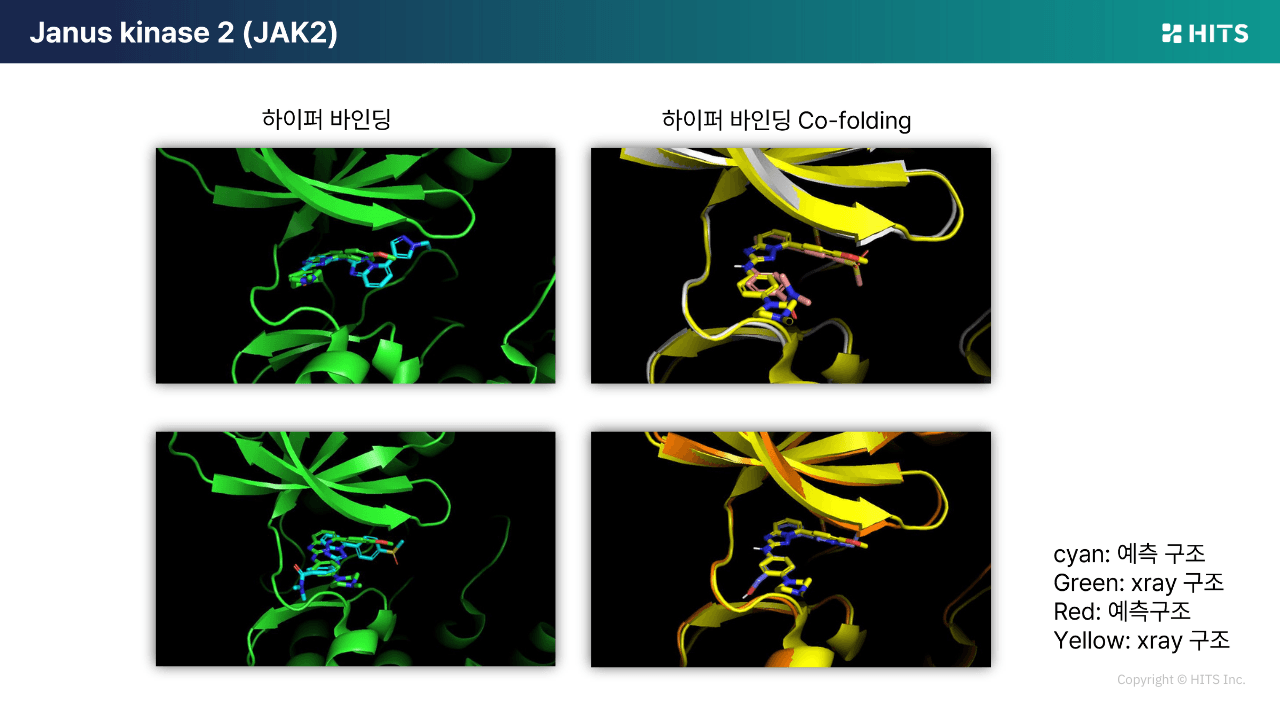
기존 도킹의 활성 예측력은 상관계수 약 0.38 수준에 머물러, 합성 우선순위를 정하는 데 한계가 있었습니다.
반면 하이퍼 바인딩 Co-folding은 동일한 세트에서 상관계수를 0.71까지 끌어올려, “합성할 후보”와 “건너뛸 후보”를 실질적으로 구분할 수 있었습니다. 또한 예측된 결합 포즈의 X-ray 정합도 역시 크게 개선되었습니다.
케이스 2. Molecular Glue 복합체
E3 ligase–표적–glue 삼자 복합체의 경우, Co-folding으로 예측한 구조가 실험 구조와 잘 일치했습니다. 더 흥미로운 점은, glue를 제거한 상태에서 예측했을 때 결합 양상이 달라졌다는 사실입니다. 이는 모델이 glue에 의해 유도되는 표면 변화를 단순한 수치 차원이 아니라, 구조적으로 이해하고 있음을 보여줍니다.
벤치마크로 알아보는 하이퍼 바인딩 Co-folding의 성능
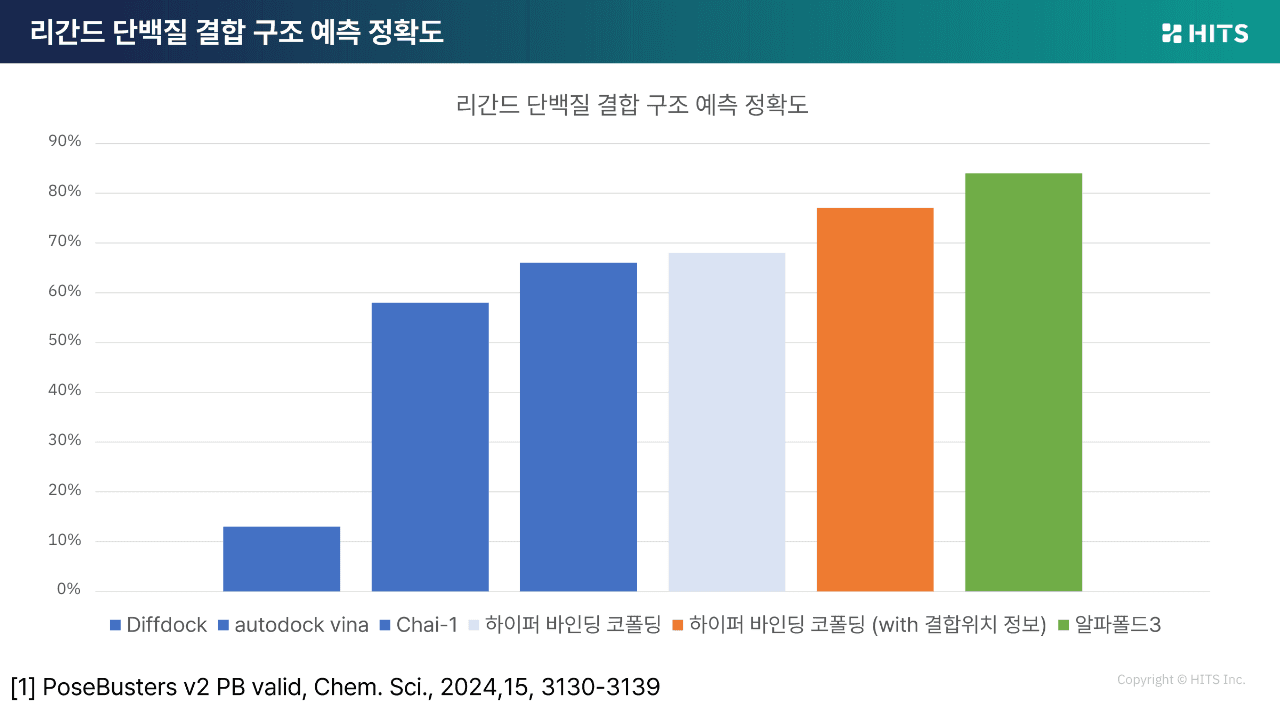
PoseBusters 기준으로 결합 구조 예측 정확도를 비교한 결과, AlphaFold 3가 약 86%로 가장 높은 성능을 보였고, 하이퍼 바인딩 Co-folding은 약 76%로 그 뒤를 이어 2위를 기록했습니다.
DiffDock, AutoDock Vina, Kai(Co-folding) 등 주요 비교군보다 높은 성능을 보여주었으며, 특히 Co-folding은 단백질 서열과 바인딩 사이트 정보만으로 이 성능을 달성했다는 점에서 주목할 만합니다.
활성 예측 정확도에서도 차이는 분명했습니다. 전통적인 도킹은 피어슨 상관계수 기준 0.1 미만으로 매우 낮은 반면, 하이퍼 바인딩(기존 도킹의 고도화 버전)은 약 0.35, Co-folding은 그 이상의 성능을 보여 실제 실험적 의사결정에 더 가까운 결과를 도출했습니다.
Co-folding의 한계점
Co-folding 역시 만능은 아닙니다. PDB에 유사한 구조가 거의 없는 특이 케이스에서는 예측이 빗나갈 수 있으며, 데이터 기반 모델의 특성상 드물게 카이랄성(chirality)이 뒤바뀌는 사례도 관찰됩니다. (다만, 이러한 문제는 후처리 과정에서 상당 부분 보완할 수 있습니다.)
또한, 큰 규모의 형태 전이와 같은 동역학적 변화를 정확히 포착하는 것은 여전히 어려운 과제로 남아 있습니다. 최근에는 MD(분자동역학) 궤적 데이터를 학습에 접목하여 이러한 한계를 극복하려는 시도들이 활발히 보고되고 있습니다.
PDB 없이도 더 정확하고, 더 빠르게
정리하자면, Co-folding은 리간드와 단백질을 함께 예측함으로써 유도 적합(induced fit)을 반영하고, PDB 없이 단백질 서열만으로 포즈를 생성하며, 바인딩 사이트 지정에 대한 부담도 줄여줍니다. 실제 벤치마크와 프로젝트 사례에서도 결합 포즈 정확도와 활성 예측력 향상이 확인되어, 신약 개발 초기 단계에서 더 빠르고 효율적인 의사결정을 가능하게 합니다.
🚀하이퍼 바인딩 Co-folding, 하이퍼랩에서 무료 체험할 수 있습니다
지금 바로 타깃을 등록하고, 직접 그 성능을 경험해 보세요.
AI 신약개발 플랫폼 하이퍼랩