Reflections on the Excellence Award at the 2nd AI Drug Discovery Competition


The 2nd AI Drug Discovery Competition: A Challenge
Hello, this is Sunghan Bae from the HITS AI Research Team.
Last July, the 2nd AI Drug Discovery Competition, hosted by the Korea Pharmaceutical and Bio-Pharma Manufacturers Association, took place. During the inaugural competition last year, I participated as part of my first project after joining HITS, working closely with my AI research team. Through constant discussions and collaboration, we steadily climbed the leaderboard, ultimately securing an impressive 4th place out of more than 700 teams.
This year, I decided to challenge myself individually, eager to assess how much I’ve grown over the past year at HITS. Once again, I was honored to achieve a 4th-place finish, receiving the Excellence Award. This recognition allowed me to attend the award ceremony and the AI Pharma Korea 2024 Conference on October 31. In this post, I’d like to share the AI drug discovery techniques I implemented during the competition and reflect briefly on my experience, in hopes that this may help a reader in the future!

How to Overcome Data Limitations?
The task for the 2nd AI Drug Discovery Competition was to build an AI model capable of predicting IC50 values—a measure of the inhibitory ability of drugs targeting IRAK4, a type of kinase involved in immune signal transduction. Participants were provided with training data consisting of IC50 values for only 1,952 compounds. This small dataset posed a significant challenge, continuing the same high level of difficulty from last year’s competition.
AI models trained on limited data often memorize patterns from the training data, leading to overfitting and poor performance on unseen test cases. To overcome these challenges, I applied the following three key strategies:
- Developing a model based on MolClr, a pre-trained Graph Neural Network (GNN).
- Addressing IC50 label imbalance using LDS (Label Distribution Smoothing) and FDS (Feature Distribution Smoothing).
- Leveraging binding affinity predictions as features to guide IC50 predictions.
Learning Molecular Data: Leveraging MolClr
First, I would like to introduce MolClr, which was the backbone for my AI model in this competition. MolClr is a pre-trained Graph Neural Network (GNN) designed specifically for molecular data. Pre-training involves teaching an AI model with data from a specific domain beforehand, equipping it with general knowledge about that domain. This enables the model to make accurate predictions with relatively small datasets while avoiding overfitting. Pre-training is particularly valuable in scientific and experimental fields, where data for individual tasks is often limited, and its adoption has been rapidly growing.
Among the prominent pre-trained models in the molecular domain, MolClr stands out. It has been pre-trained on over 10 million molecules from PubChem, the largest chemical database. Using a method called contrastive learning, MolClr distinguishes between real molecules from PubChem and artificially altered molecules, where atoms, bonds, or functional groups are randomly obscured or removed. Through this process, MolClr learns the structural and chemical properties inherent to molecules. This pre-trained knowledge compensates for the lack of structural and chemical diversity in the training data, mitigating overfitting risks during the model training process.
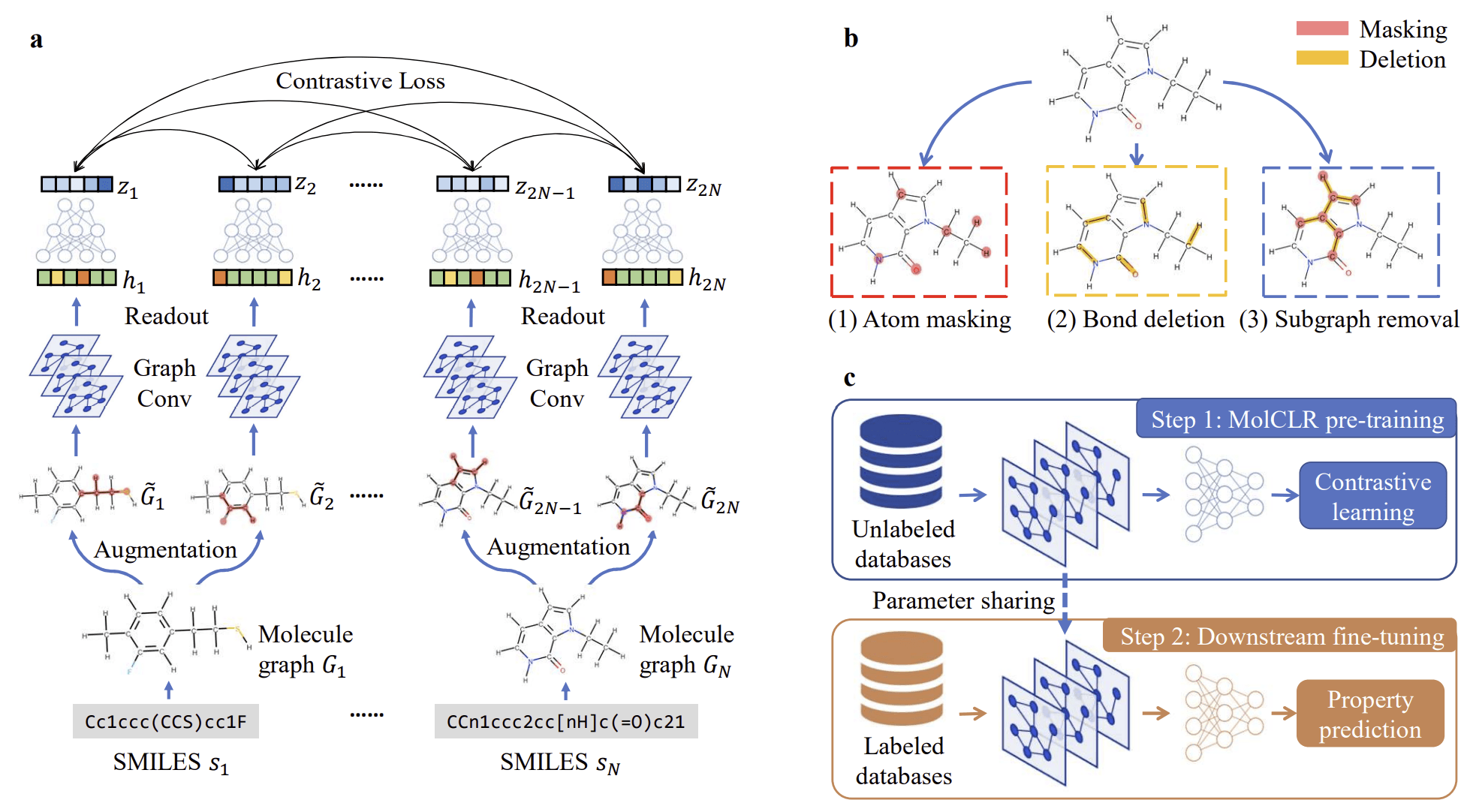
Another strategy I used to prevent overfitting was addressing label imbalance in the training data. Imbalance in IC50 values can significantly impact the model's performance, as the diversity in molecular structure and chemistry alone may not be enough to achieve accurate predictions. For instance, if IC50 values are predominantly distributed in the lower range, a model trained on such data may fail to predict higher IC50 values effectively. To address this imbalance, I incorporated LDS (Label Distribution Smoothing) and FDS (Feature Distribution Smoothing) techniques, ensuring the model could make more balanced and accurate predictions for unseen data.
Overcoming Imbalance with LDS and FDS
LDS (Label Distribution Smoothing) and FDS (Feature Distribution Smoothing) are deep learning techniques proposed in the 2021 paper Delving into Deep Imbalanced Regression. These methods are designed to address label imbalance in regression tasks where continuous labels are unevenly distributed. Such imbalances can lead to biased predictions by AI models. As their names suggest, both techniques smooth the data distribution: LDS focuses on the label distribution, while FDS targets the feature distribution that the deep learning model uses during training.
In essence, LDS and FDS approximate the imbalanced data with a smoother distribution, assuming that the smoothed feature and label distributions align closely. This adjustment helps the model generalize better, reducing overfitting caused by label imbalance. Implementing LDS and FDS in my model significantly narrowed the gap between training and test performance, which led to one of my two key "jump-ups" during the competition.
The other jump-up came from incorporating inductive bias into my model. Inductive bias refers to assumptions embedded in a model that improves its ability to learn specific tasks. A prime example of inductive bias is the GNN (Graph Neural Network) structure of the MolClr model, which mimics the relationships within graph-shaped data such as molecules. A GNN leverages the graph's nodes (e.g., atoms) and edges (e.g., bonds) to analyze patterns effectively. This inductive bias makes GNNs particularly well-suited for learning and understanding molecular data, cementing their widespread use in this domain.
Finding Clues for IC50 with Binding Affinity
One of the inductive biases I introduced in my model during the competition was leveraging predicted binding affinity energy values as features. To understand why this approach was effective, let’s first consider the concept of IC50. IC50 refers to the concentration of a drug required to inhibit 50% of the activity of a target protein (in this task, IRAK4). It is a quantitative measure obtained by plotting a dose-response curve after applying the drug to the target protein. In essence, IC50 is a macroscopic indicator of a drug’s inhibitory ability within the context of an entire system, such as cells or tissues.
On a microscopic scale, however, the mechanism of drug inhibition involves the drug molecule chemically binding to a specific site on the target protein. The strength of this binding interaction is referred to as binding affinity. Thus, we can hypothesize that the microscopic binding affinity between a drug and its target protein strongly correlates with the macroscopic inhibitory metric, IC50. Binding affinity, therefore, serves as a useful clue—or inductive bias—for predicting IC50 in this task.
Normally, obtaining binding affinity values would require labor-intensive experimental measurements, making it challenging to directly use them as features for AI model training. However, by employing AI-predicted binding affinity values, it becomes feasible to utilize this crucial information in a computational framework.
Hyper Lab: The Foundation for Progress
Thanks to Hyper Lab, HITS’s AI-driven drug discovery platform, it becomes possible to quickly and efficiently predict the binding structure and energy of drug molecules within target proteins, using only their structural information. HITS’s AI models for drug-protein binding prediction are designed with advanced network structures and algorithms that mimic molecular interactions at the microscopic level, delivering exceptionally high accuracy.
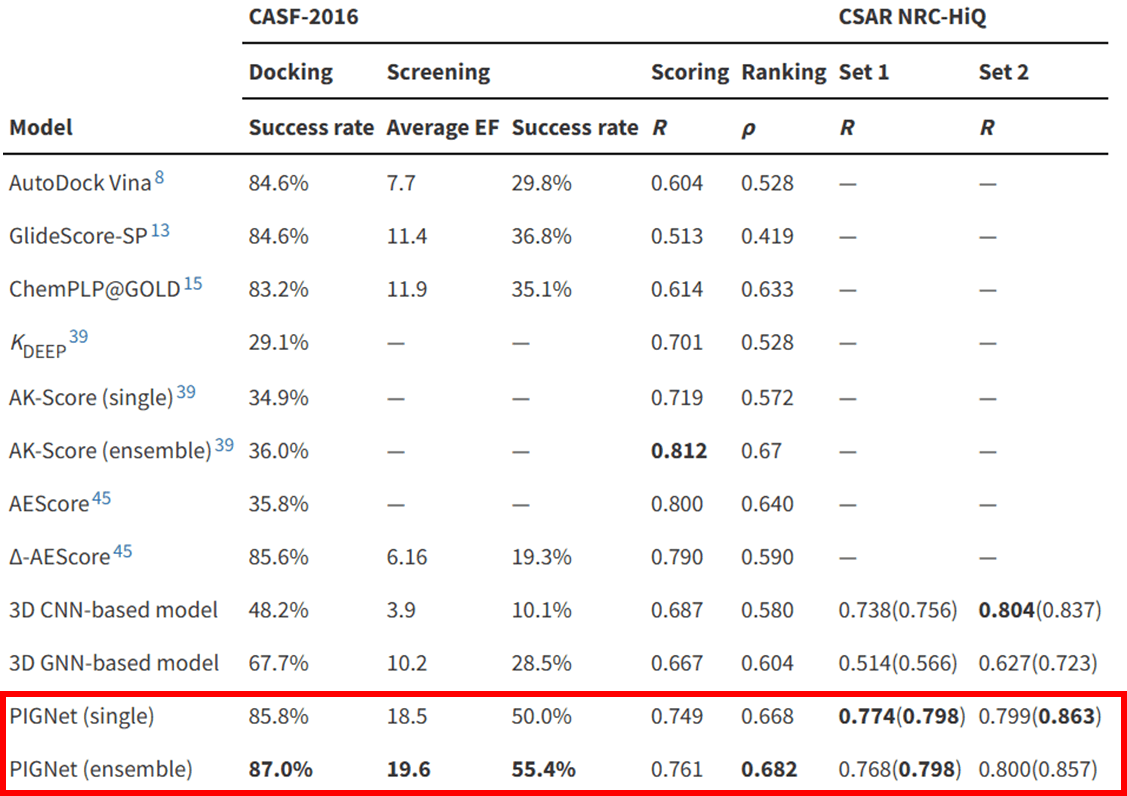
For instance, Hyper Lab’s binding affinity prediction module, PIGNet, demonstrates outstanding performance in binding affinity benchmarks like CASF-2016, achieving a Pearson correlation coefficient of approximately 0.77. This enables us to obtain reliable, AI-predicted binding affinity values for large datasets of drugs in a fraction of the time it would take to conduct laboratory experiments. These predictions can then be used as inductive bias features for IC50 preditions.
During the competition, incorporating binding affinity predictions generated by Hyper Lab into my model resulted in a significant "jump-up" in the leaderboard rankings. This highlights Hyper Lab's ability to address experimental challenges and streamline the AI-driven drug discovery process.
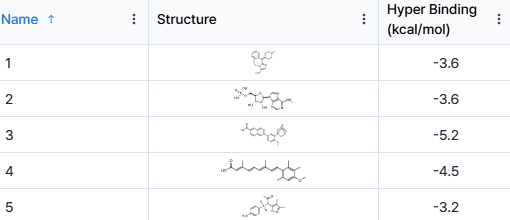
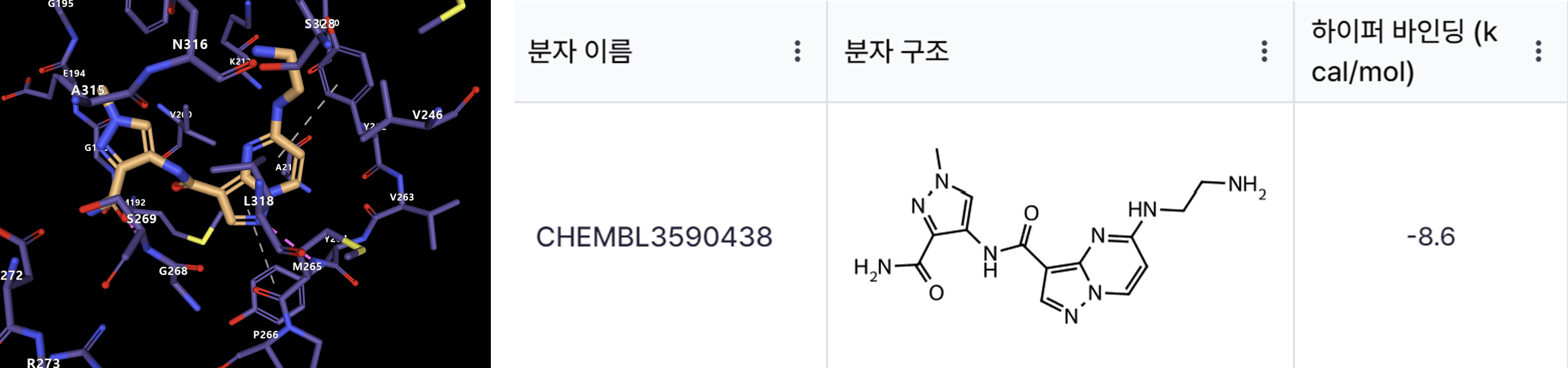
Continuing Challenges and Growth with HITS
In this post, I have shared the various AI drug discovery techniques I applied during the competition. Last year, I entered the inaugural competition as a newcomer to this field, participating in my first project. This year, however, I had the opportunity to leverage the experience and insights I gained over the past year, achieving an award-winning result that I am deeply proud of.
I tackled challenges such as the limitations of the training data—its scarcity and label imbalance—while gaining a deeper understanding of the task, including the relationship between IC50 and binding affinity. Formulating hypotheses to address these issues and conducting experiments that led to significant leaderboard improvements was an incredibly rewarding experience as a researcher. Furthermore, utilizing HITS’s innovative technology to predict binding affinity, which is otherwise difficult to use as an AI feature due to its experimental nature, filled me with immense pride in our capabilities.
My journey of challenges and growth does not stop here. I will continue striving to advance, contributing to HITS’s innovations, and working towards making the world a better place. Together with HITS, I am committed to creating meaningful, positive change through technology.