Virtual Library와 Similarity Search


Virtual Library란?
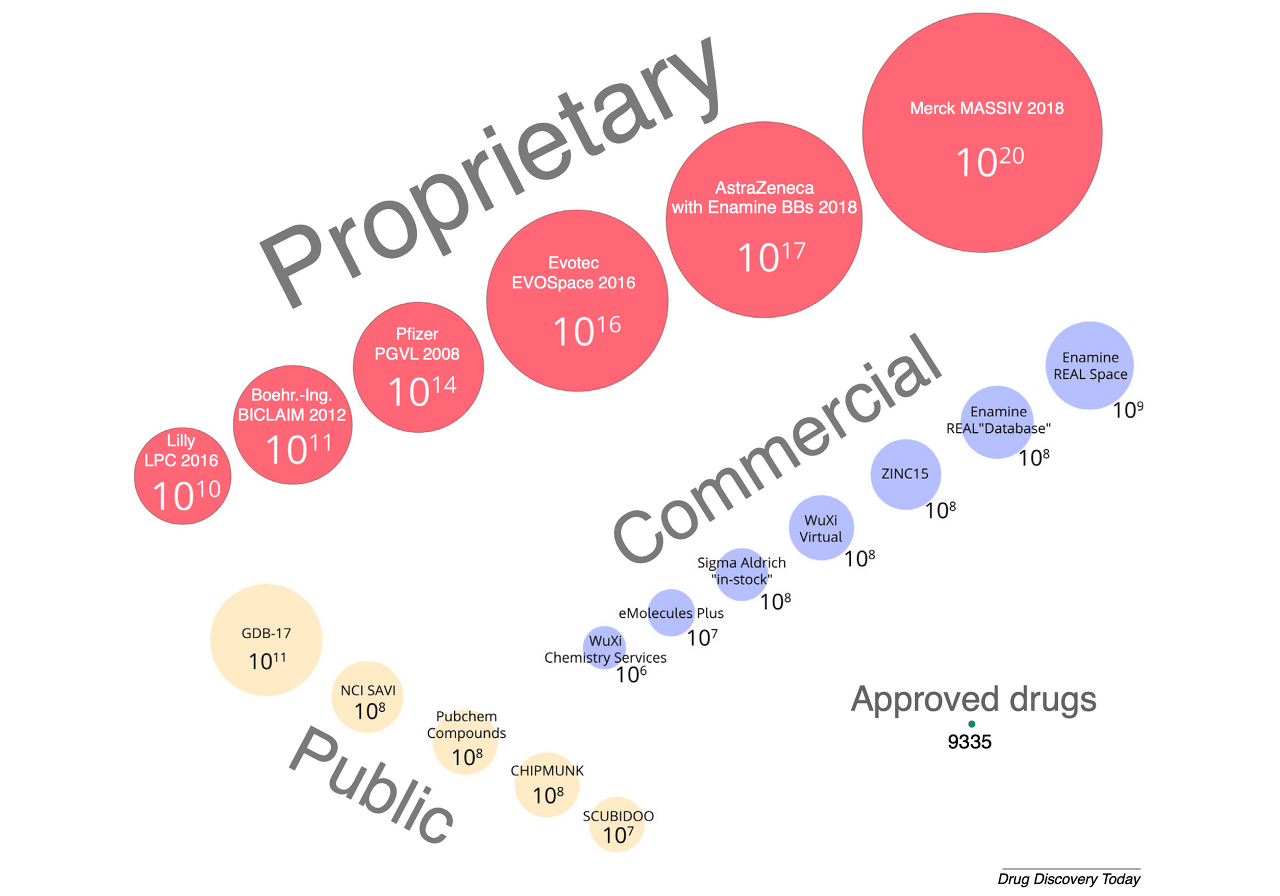
Virtual Library란, 약물이 될 가능성이 있는 다량의 가상 화합물을 컴퓨터라는 저장 공간에 저장해 놓은 데이터를 의미합니다. Virtual Library는 일종의 잠재적인 약물 후보군과도 같기 때문에, 최종적으로 선발될 가능성이 조금이라도 높은 후보들을 최대한 많이 확보하는 것이 중요합니다. 이 때문에 Virtual Library는 대개 그 크기가 상상할 수 없을 정도로 큽니다. 실제로 글로벌 기업들이 보유한 Virtual Library 중에는 관측 가능한 우주에 존재하는 별의 개수(10¹¹)보다 더 많은 경우도 있습니다. 실용적인 Virtual Library를 만들기 위해서는 필요 시 쉽게 합성할 수 있는 분자들로 구성하는 것이 매우 중요합니다.
Virtual Library를 만드는 방법
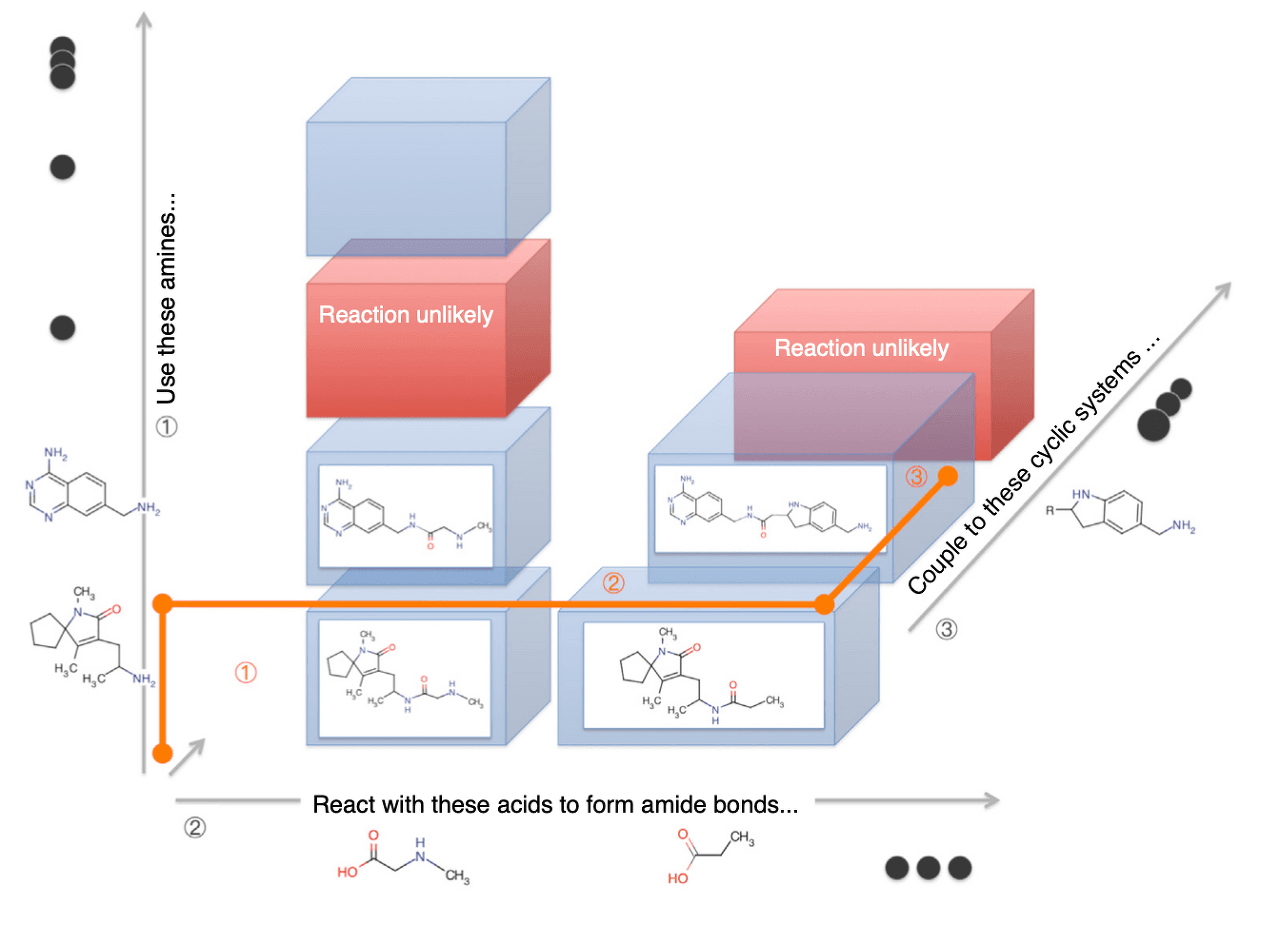
천문학적인 숫자의 분자로 구성된 Virtual Library를 사람이 하나하나 설계하는 것은 사실상 불가능합니다. 합성 용이성(synthesizability)과 drug-likeness(약물 유사성)도 함께 고려해야 하기 때문에, 규칙성 없이 무작위로 원자들을 이어 붙이는 방식은 매우 비효율적입니다. 이 때문에 통상적으로 Virtual Library를 생성할 때는 다음과 같은 방법들이 사용됩니다.
먼저 Building Block이라고 할 수 있는 크기가 작은 화합물의 Pool을 생성합니다. 이 Pool에 속하는 화합물은 산업적으로 합성이 용이해야 합니다. 화합물의 약물 가능성이 높다고 하더라도 합성 생성물의 수율이 낮거나 그 비용이 비싸다면 상업성이 매우 떨어지기 때문입니다. 그리고 해당 Building Block들끼리 일어날 수 있는 화학 반응을 적용하여 새로운 화합물을 만들어냅니다. 이 과정에 이용되는 화학 반응은 매우 단순한 과정으로 손쉽게 일으킬 수 있는 화학 반응이어야 합니다. 이제 새로 생성된 화합물을 Building Block으로 삼아 위 과정을 반복하면 합성이 용이한 화합물을 대량으로 생성할 수 있습니다.
Virtual Library를 탐색하는 방법
Virtual Library를 탐색하는 것은 상당한 시간이 소요되는 작업입니다. 신약 개발의 초기 후보물질 발굴 과정은 docking이라는 일련의 계산 과정을 필요로 합니다. 이 과정은 계산량이 매우 많아 긴 시간이 소요됩니다. 예를 들어, Virtual Library에 포함된 화합물의 개수가 40억 개에 달하고, 이들 모두에 대해 Docking을 수행해야 한다면, 100개의 CPU로 약 3년이 걸립니다. 수개월 내에 이 작업을 완료하려면 수천 개의 CPU가 필요하며, 이는 통상적인 계산 자원으로는 감당하기 어렵습니다. 이 지점에서 합리적인 Virtual Library 탐색 전략의 필요성이 대두됩니다.
Similarity Search
Similarity Search는 유사한 구조를 가지는 화합물은 같은 타겟 단백질에 대해 유사한 활성을 지닐 것이라는 가정에 근거한 탐색 방법입니다. 활성이 있는 것으로 알려진 화합물과 유사한 구조를 지니는 화합물을 Virtual Library에서 찾을 수 있다면, 이 유사한 구조의 화합물만을 선별해 Docking을 수행함으로써 계산 시간을 크게 단축할 수 있을 것입니다. 또는 소수의 분자에 대해서만 Docking 계산을 수행한 뒤, score가 높은 물질을 선별하고, 그 물질과 유사한 구조의 화합물에 대해서만 추가적으로 Docking 계산을 수행할 수도 있습니다. 물론 이러한 Similarity Search 과정은 Docking 과정보다 훨씬 빠르게 수행되어야 효율성이 있겠죠. 그럼 이제 서로 다른 두 화합물이 얼마나 유사한지를 계산하는 방법에 대해 알아보겠습니다.
Molecular Fingerprint
두 화합물의 유사도를 논하려면 먼저 '무엇이' 유사한지를 결정해야 합니다. 즉, 어떤 기준으로 두 화합물을 비교할 것인지를 정의해야 하죠. 이를 위해 컴퓨터에게 분자의 특성을 설명해 주어야 하는데, 이때 사용되는 것이 바로 Molecular descriptor입니다. Molecular Fingerprint는 이러한 Molecular descriptor 중 가장 보편적으로 사용되는 개념으로, '분자 구조'라는 정의하기 어려운 추상적인 개념을 유한한 차원의 벡터(vector)로 표현하는 방식입니다. 즉, 분자의 구조를 컴퓨터가 이해할 수 있도록 수치화한 형태라고 할 수 있습니다.
Structural key based Fingerprint
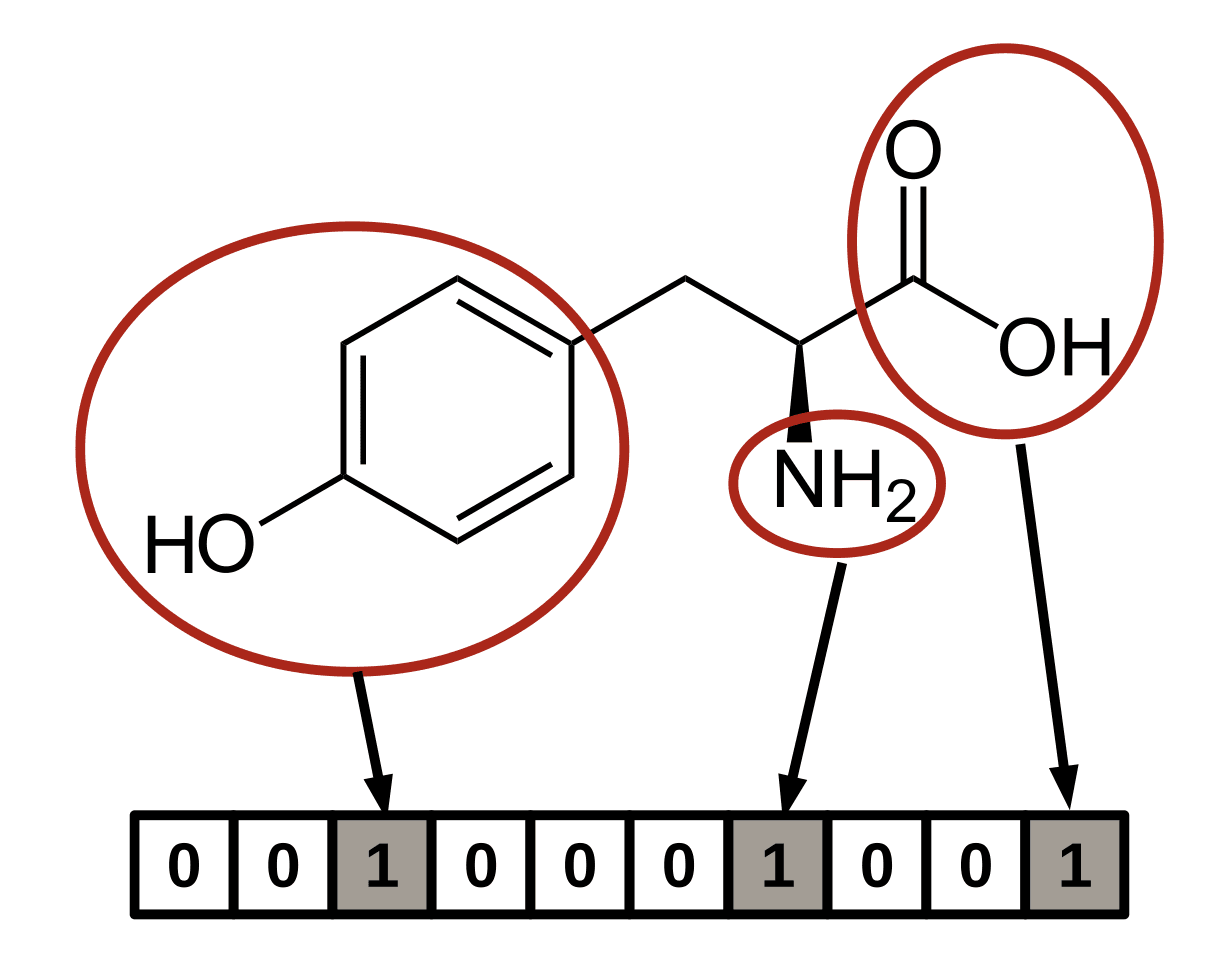
이 Fingerprint는 미리 정해놓은 화학 작용기 혹은 특정 원자가 분자 내에 존재하는지를 각 비트(bit)에 저장해 벡터 형태로 표현합니다. 예를 들어, 아래 그림은 화합물에 Phenyl group이 존재할 경우 Fingerprint 벡터의 세 번째 요소를 1로, 존재하지 않을 경우에는 0으로 지정하는 규칙을 보여주는 예시입니다.
이처럼 관심 있는 화합물이 특정 조건을 만족하는지를 명시적으로 표현하는 데에는 매우 적합한 방식입니다. 그러나 이러한 방식에는 한 가지 단점도 존재합니다. 바로 해당 작용기나 원자들이 분자 내에서 어떻게 배치되어 있는지, 즉 위치 관계(공간적 배열)에 대한 정보는 Fingerprint에 포함되지 않는다는 점입니다.
Hashed Fingerprints
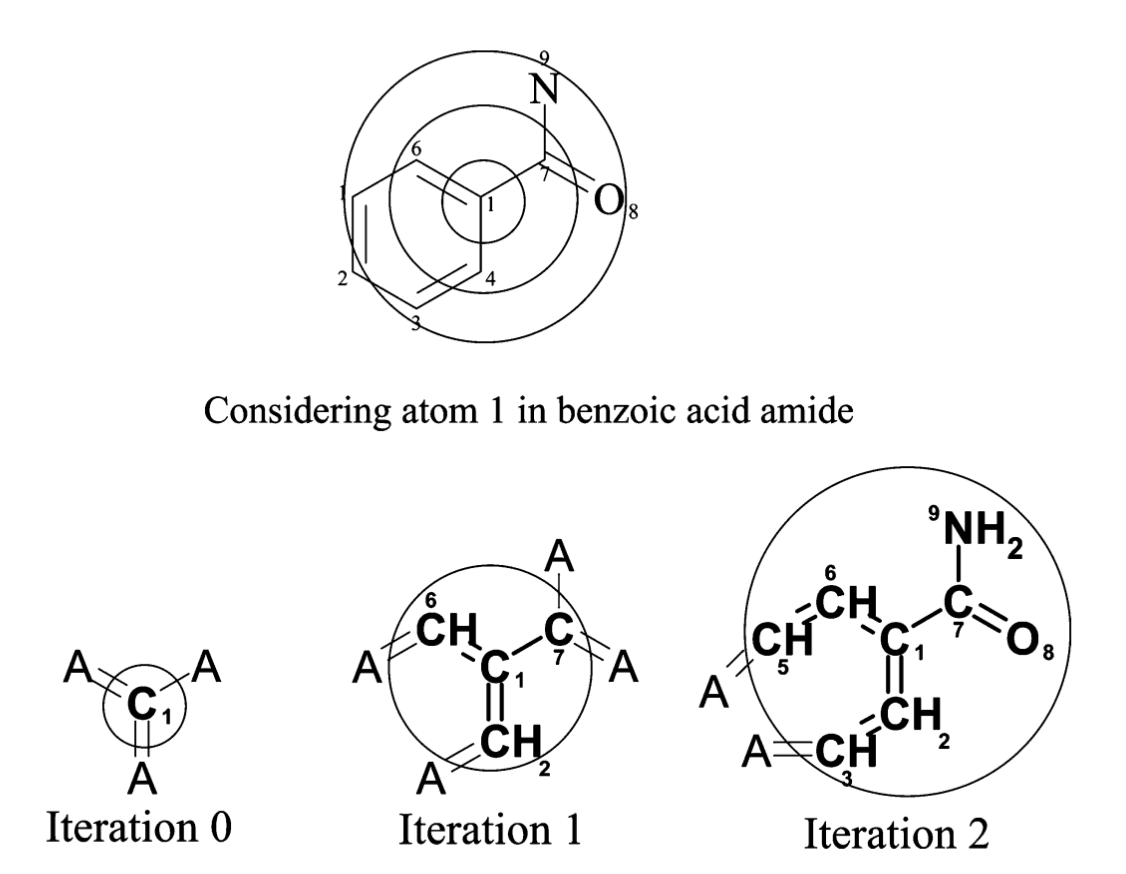
여기서는 Hashed Fingerprint 중에서 Circular Fingerprint의 한 종류인 ECFP(Extended-Connectivity Fingerprints)를 소개합니다. ECFP를 계산하기 위해서는 먼저 원자 간 거리와 Fingerprint의 bit 수를 미리 설정해야 합니다.
ECFP는 여섯 개의 기준을 이용해 각 원자에 대한 특성을 1차적으로 계산합니다. 여기서 원자 간 거리란, 아래 그림처럼 동심원의 중심에 위치한 원자를 기준으로 인접한 원자를 몇 개까지 탐색할 것인지에 대한 값입니다. 이와 같은 방식으로 화합물 내 수소를 제외한 모든 원자를 순회하면서, 앞서 언급한 여섯 개의 기준에 대한 특성을 얻어냅니다. 그런 다음 이 정보를 Hash 함수를 이용해 사용자가 설정한 bit 수에 맞춰 정보를 저장합니다. 이렇게 생성된 결과는 Structural keys 기반의 Fingerprint와 마찬가지로 0과 1로 구성된 벡터로 표현됩니다.
Molecular Similarity
이제 거의 다 왔습니다. 지금까지 각 화합물의 유사도를 얻기 위해, 각 화합물의 특성을 수학적으로 기술하는 방법에 대해 알아보았는데요. 이제는 이 각각의 기술자(Descriptor)가 서로 얼마나 유사한지를 계산할 차례입니다. 우리는 Similarity를 계산하는 방법 중 하나인 Tanimoto Coefficient에 대해 알아보겠습니다. 이 방법은 앞서 구한 벡터들의 각 element를 같은 순서대로 비교하여, 일치하는 bit 수와 다른 bit 수를 계산하고, 이를 바탕으로 유사도를 정량화합니다. 구체적인 수식은 다음과 같습니다.
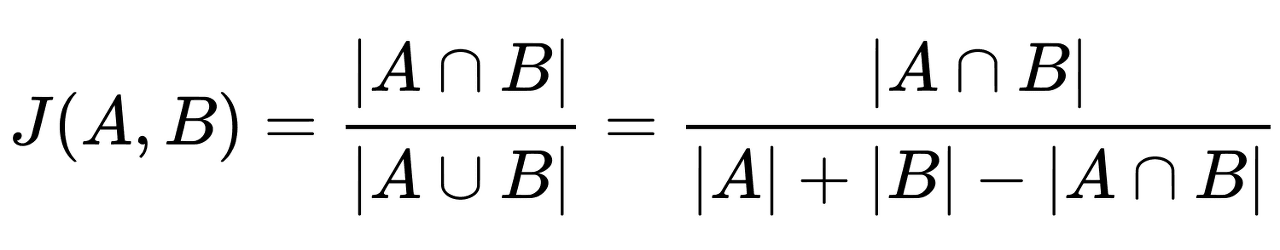
위와 같은 수식을 적용하면, 서로 다른 두 분자의 유사도가 0에서 1 사이의 값으로 계산됩니다. 유사도 값이 1에 가까울수록 두 분자의 구조적 유사성이 높다는 것을 의미하며, 두 분자가 완전히 동일할 경우 유사도는 1이 됩니다. 이러한 방식으로 분자 간의 유사도를 정의하고, 그 유사도에 따라 Virtual Library에서 '유사한' 분자들을 선별함으로써, 대규모 Virtual Library를 보다 효율적으로 탐색할 수 있습니다.