ICML 2024 미리 보기


ICML이란
지난 ICLR 2024 미리 보기에서 세계 3대 인공지능 학회로 ICLR, ICML, NeurIPS를 소개해드렸습니다. 얼마 전 5월 오스트리아 빈에서 ICLR 2024이 진행되었는데요, 다가오는 7월에 같은 장소에서 제 41회 ICML, The 41st International Conference on Machine Learning이 개최될 예정입니다.

ICLR에 비해 ICML이 기계학습 전 분야를 보다 폭넓게 다룬다는 의견도 있지만, 실제 발표된 연구들을 보면 두 학회 간의 차이는 비교적 크지 않은 것으로 보입니다. ICLR처럼 ICML도 학회 개최에 앞서 승인된 논문들이 미리 공개되었습니다.
지난 글에 이어 이번에는 ICML 2024에서 승인된 논문 중 신약개발 관련 딥러닝 연구들을 모아보고 그 중 5건의 연구들을 한 번 살펴보겠습니다.
ICML 2024에 발표되는 신약개발 관련 분야 연구들
이번에도 승인 논문 목록에서 chem, bio, molecule, drug, protein 등 관련 키워드를 통해 신약개발과 관련된 연구들을 추려보았습니다. 그렇게 약 60 건의 논문을 모으고 주제를 나눠보았습니다.
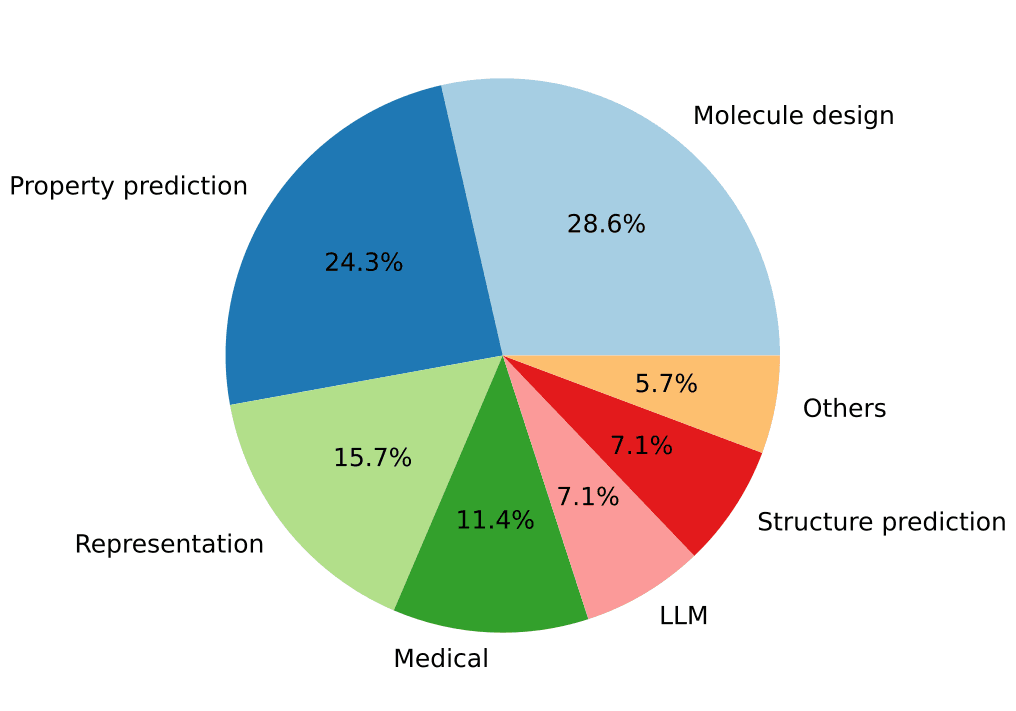
ICLR 2024 때와 마찬가지로, 이번에도 분자를 설계하거나(즉, 분자 생성 AI를 개발하거나) 분자 시스템의 다양한 물성을 예측하는 연구가 주를 이뤘습니다. 분자 표현 학습(representation learning) 연구는 대부분 물성 예측을 목적으로 하기 때문에, 이 두 카테고리의 연구는 상당 부분 중첩됩니다. 그 외에는 의학 분야의 문제를 다룬 연구, 거대언어모델(large language models, LLMs)을 화학·생명과학 문제에 적용한 연구, 분자 시스템의 구조를 예측하는 연구 등이 뒤를 이었습니다.
위 통계에 포함된 논문들의 총 목록은 아래와 같습니다.
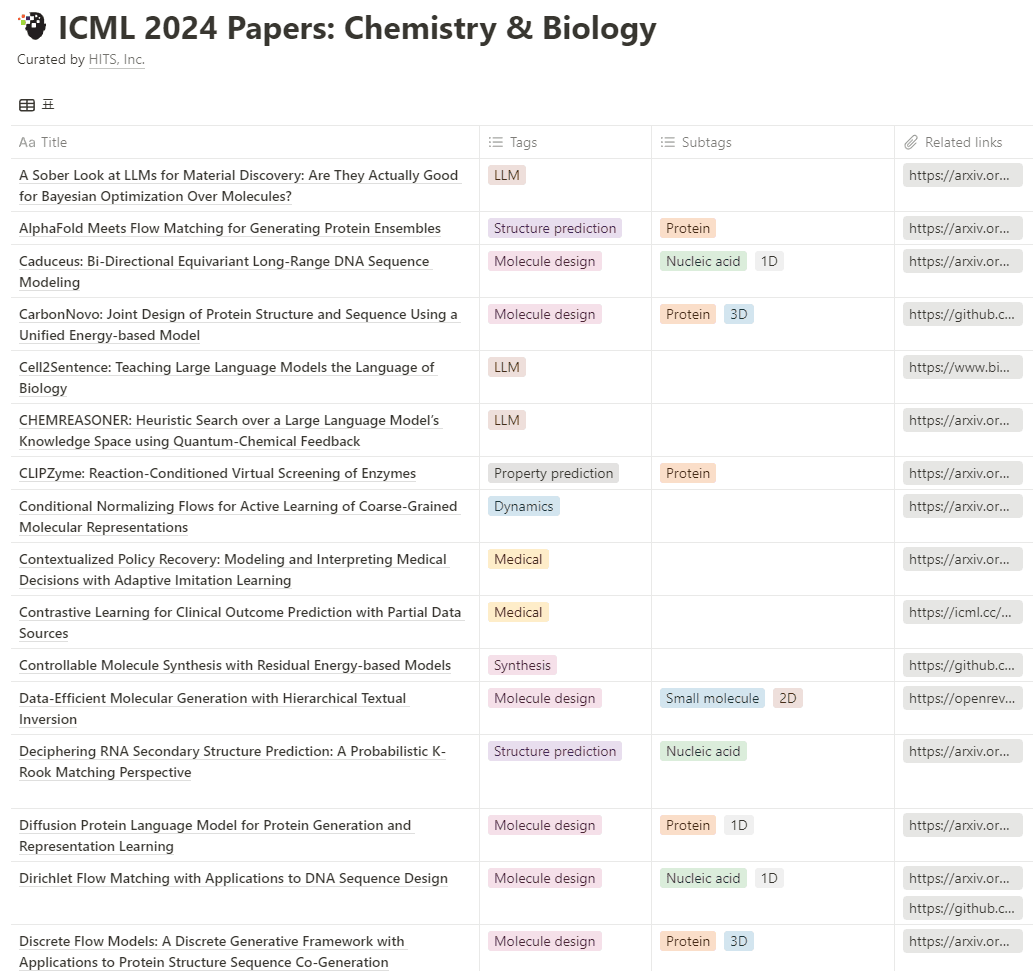
그럼 어떤 연구들이 발표되었는지 좀 더 자세히 알기 위해 위 목록으로부터 다음의 5개 ICML 2024 논문들을 뽑아 하나씩 살펴보겠습니다. 참고로 앞서 보여드린 주제별 통계와는 무관하게 각 논문에서 다루는 문제에 주안을 두고 뽑아보았습니다.
- AlphaFold Meets Flow Matching for Generating Protein Ensembles [Jing et al. ICML 2024]
- CLIPZyme: Reaction-Conditioned Virtual Screening of Enzymes [Mikhael et al. ICML 2024]
- Drug Discovery with Dynamic Goal-aware Fragments [Lee et al. ICML 2024]
- MolCRAFT: Structure-Based Drug Design in Continuous Parameter Space [Qu et al. ICML 2024]
- A Sober Look at LLMs for Material Discovery: Are They Actually Good for Bayesian Optimization Over Molecules? [Kristiadi et al. ICML 2024]
ICML 2024 논문 미리 보기 1 - 단백질의 다양한 3차원 구조 예측하기
단백질의 3차원 구조는 고정되어 있지 않고, 시간에 따라 유동적으로 변한다는 사실은 잘 알려져 있습니다. 이는 단순한 열운동을 의미하기보다는, 단백질의 안정된 구조(conformation)가 환경에 따라 달라질 수 있다는 뜻입니다. 실제로 Protein Data Bank(PDB)에 동일한 단백질의 서로 다른 X-ray 구조들이 다수 등재되어 있는 것만 봐도 이를 알 수 있죠.
AlphaFold의 놀라운 성능이 발표되었을 때, 이미 많은 연구자들이 이 모델이 단일한 단백질 구조만을 예측한다는 점을 한계로 지적했습니다. 이후 AlphaFold와 같은 모델을 활용해 단백질의 다양한 conformation, 즉 구조의 ensemble을 예측할 수 있는 방법에 대한 연구가 이어졌습니다.
AlphaFold로 단백질 구조의 ensemble을 예측하는 대표적인 방법으로는 MSA subsampling이 있습니다. MSA(Multiple Sequence Alignment, 다중서열정렬) 데이터는 AlphaFold와 같은 모델이 단백질 서열의 3차원 구조를 예측할 때 중요한 참고 자료로 사용됩니다. 이는 진화적 관계에 기반한 서열 간 동종성을 유추하는 데 도움을 줍니다.
MSA subsampling이란, 전체 MSA 데이터 중 일부만을 선택해 AlphaFold가 사용하도록 하는 방법입니다. 서로 다르게 추출된 MSA 샘플들이 동일한 단백질의 서로 다른 conformation을 예측할 수 있게 해준다는 연구 결과들이 보고되어 왔습니다. [Wayment-Steele et al. Nature 625: 832 (2024)]

이번 ICML 2024의 연구 “AlphaFold Meets Flow Matching for Generating Protein Ensembles”는 동일한 문제를 전혀 다른 방법으로 해결합니다. [Jing et al. ICML 2024] AlphaFold는 주어진 단백질 서열에 대해 최적의 단일 구조를 예측하는 AI입니다. MSA subsampling은 이 같은 예측 AI가 동일한 질문 (서열)에 대해 서로 다른 예측값을 내놓게 하는 일종의 트릭인 셈이죠. [Jing et al. ICML 2024]에서는 MSA subsampling에 의존하는 대신 AlphaFold를 생성 AI로 바꾸는 방법을 제시합니다. 다양한 결과를 내놓을 수 있는 생성 AI의 본질을 자연스럽게 이용하여 단백질 ensemble을 예측하는 것이죠.
이 때 생성 모델의 형태 및 학습 방법으로 최근에 각광받는 flow matching [Lipman et al. ICLR 2023]을 사용했습니다. 또한, MSA 데이터를 조작할 필요가 없기 때문에 ESMFold [Lin et al. Science 379: 1123 (2023)]처럼 애초에 MSA 데이터를 사용하지 않는 단백질 구조 예측 모델에도 적용할 수 있습니다. [Jing et al. ICML 2024]에선 AlphaFold와 ESMFold를 각각 생성 AI로 변형하여 AlphaFlow와 ESMFlow로 명명하고, 해당 모델들로 단백질 구조의 ensemble을 예측했습니다.
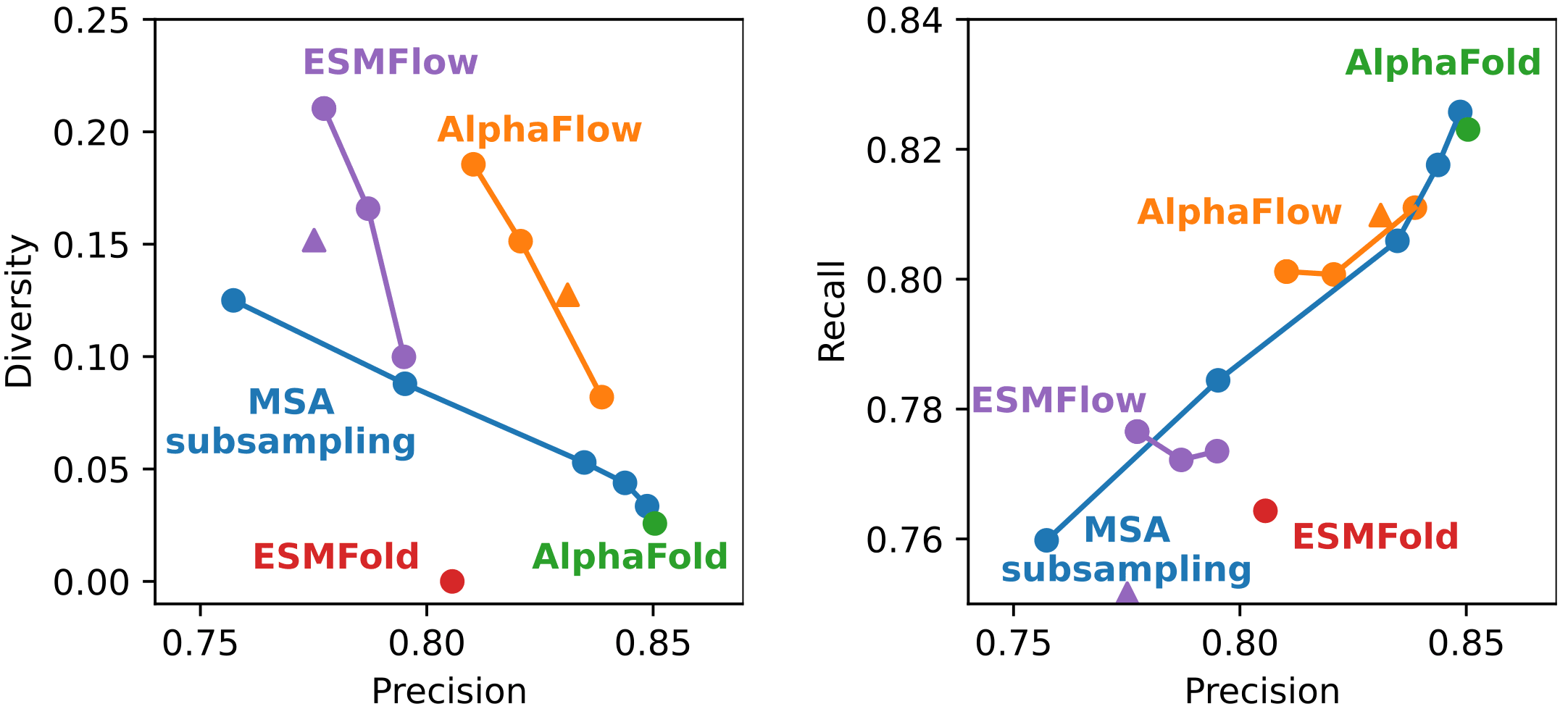
아래 그림은 AlphaFlow, ESMFlow 및 비교 모델들이 단백질 구조의 ensemble을 얼마나 잘 예측했는지를 보여줍니다. 단일 구조만을 예측하는 AlphaFold와 ESMFold는 구조 다양성(diversity)이 매우 낮거나 아예 없는 반면, AlphaFlow와 ESMFlow는 MSA subsampling보다 더 높은 다양성을 보였습니다. 세 모델 모두 추론 파라미터를 조절해 다양성을 낮추는 대신 정확도(precision)를 높이거나, 그 반대로 조정할 수 있습니다. 다만, 동일한 단백질에 대해 서로 다른 PDB 구조들을 얼마나 많이 예측해낼 수 있는지를 나타내는 recall 측면에서는, AlphaFlow와 ESMFlow 모두 추론 파라미터를 조절해도 큰 변화는 없었습니다.
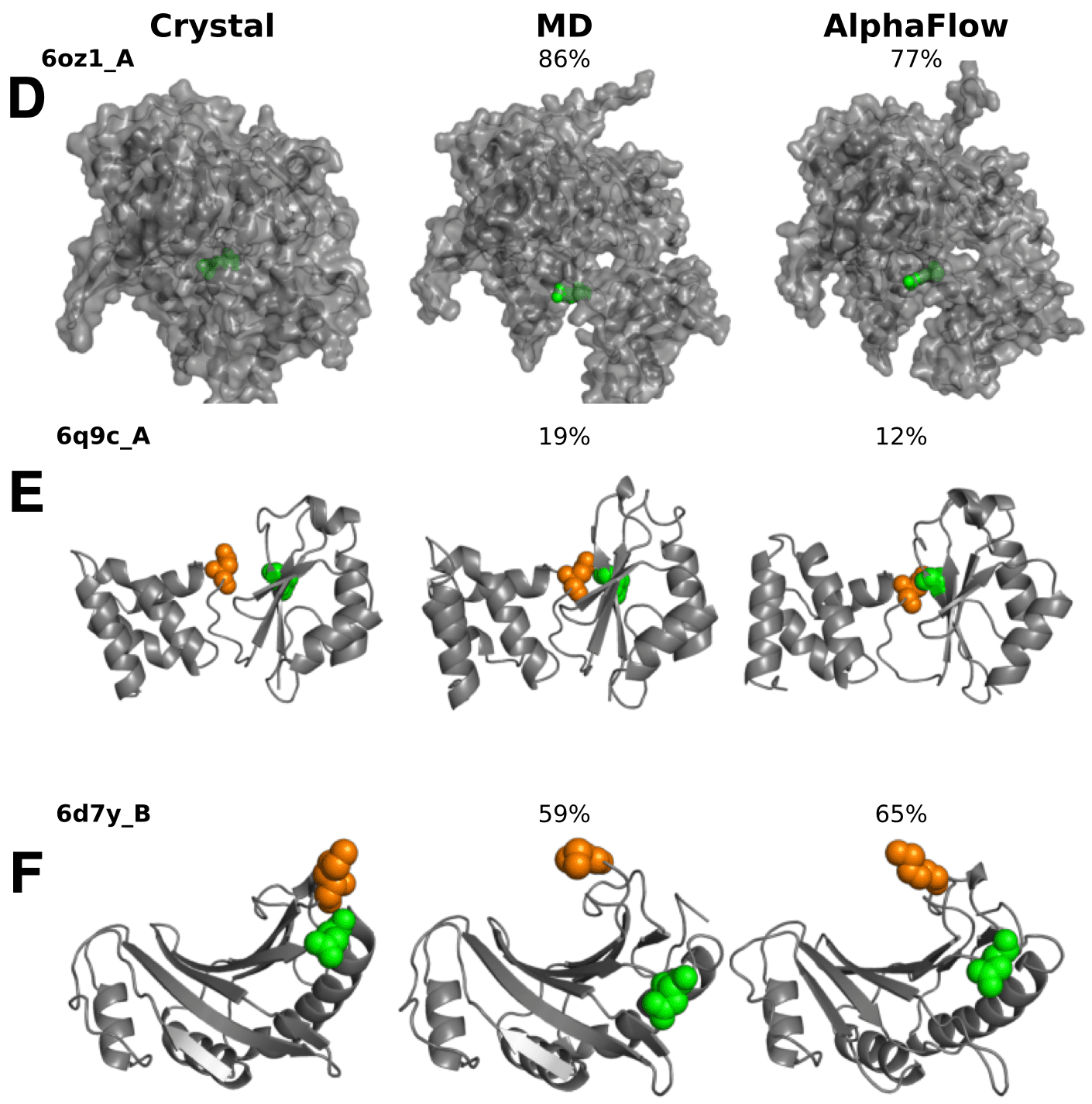
다음으로는 분자동역학(Molecular Dynamics, MD) 시뮬레이션을 통해 단백질 conformation의 변화를 모사하고, AlphaFlow가 이러한 conformation을 올바르게 예측할 수 있는지를 실험했습니다. 아래 그림과 같은 사례에서는 AlphaFlow가 PDB 결정 구조에는 포함되지 않은 또 다른 conformation을 정확하게 예측했으며, 해당 conformation의 존재 확률 또한 MD 시뮬레이션 결과와 유사하게 예측하는 것을 확인할 수 있습니다.
최근 AlphaFold 3 [Abramson et al. Nature (2024)]와 RoseTTAFold All-Atom [Krishna et al. Science 384 (2024)]처럼 단백질뿐 아니라 모든 생분자 시스템의 구조를 예측할 수 있는 놀라운 모델들이 개발되었습니다. 두 모델 모두 최적의 단일 구조를 예측하는 모델로, 위와 같은 방식으로 단백질 구조의 ensemble을 예측하는 연구는 앞으로도 계속 이어질 것으로 보입니다.
ICML 2024 논문 미리 보기 2 - 원하는 화학 반응을 촉매하는 효소 찾아내기
실험적으로 활성을 확인한 화합물의 표적을 가상으로 조사하는 방법으로 reverse docking 또는 inverse docking이 있습니다. Docking이 특정 표적 단백질에 대해 화합물의 상호작용을 예측하는 방식이라면, reverse docking은 정해진 화합물을 기준으로 유의미한 상호작용을 보일 수 있는 단백질을 탐색하는 방식입니다. 이처럼 원하는 화합물과 상호작용할 수 있는 단백질을 찾는 것과 유사하게, 특정 화학 반응을 촉매할 수 있는 효소 단백질을 탐색할 수도 있습니다. 예를 들어, 해당 화학 반응을 생합성 경로로 구현하고자 할 때 유용하죠.
거대한 화학공간에서 약물을 찾아내는 게 어려운 것처럼 원하는 효소를 찾아내는 일도 쉽지 않습니다. UniProt Knowledgebase (UniProtKB)에 등록된 2.4억 개 단백질들 중, 20%도 채 되지 않는 단백질만이 효소의 촉매 반응을 분류하는 Enzyme Commission (EC) number 번호를 가지고 있습니다. [Ribeiro et al. Biochem. J. 480: 1845 (2023)]. 아울러 작은 변화가 활성에 큰 변화를 주거나, 구조적으로 다른 효소가 동일한 화학 반응을 촉매하는 경우도 있어 예측을 더욱 어렵게 만듭니다.
최근에는 단백질의 서열로부터 EC number를 예측하는 CLEAN (contrastive-learning-enabled enzyme annotation)이라는 비교적 간단한 아이디어의 모델이 Science 지에 발표되기도 했습니다. [Yu et al. Science 379: 1358 (2023)]
이번 ICML 2024에 발표되는 “CLIPZyme: Reaction-Conditioned Virtual Screening of Enzymes”에서는 원하는 화학반을 촉매할 수 있는 단백질을 예측합니다. [Mikhael et al. ICML 2024] CLEAN 모델의 대상이 효소로부터 화학반응을 맞추는 docking과 같다면, 본 연구의 모델 CLIPZyme의 대상은 화학반응으로부터 효소를 맞추는 reverse docking과 같은 셈이죠.
CLIPZyme은 CLEAN과 비슷하게 대조 학습 (contrastive learning)을 사용하지만, 기질과 생성물 구조를 입력으로 받는다는 점과 조사할 단백질의 3차원 구조를 모델 내부에서 활용한다는 점이 다릅니다. 참고로 대조 학습이란 짝을 지었을 때 정답인 쌍들과 오답인 쌍들의 데이터를 모으고 구성한 후, 임의의 쌍이 정답인지 아닌지를 판별하도록 모델을 학습시키는 방법입니다.[Radford et al. ICML 2021] 효소 탐색처럼, 특정 화학 반응을 촉매할 수 있는 단백질과 그렇지 않은 단백질이 명확히 구분되는 문제에 적합한 방법이죠.
CLIPZyme은 화학반응에 있어서 전이상태가 중요하다는 점, 특히 효소의 활성부위 구조가 기질의 전이상태 구조를 안정화하도록 형성된다는 점에 초점을 맞춥니다. 따라서 기질과 생성물의 화학구조로부터 가상의 전이상태 구조(pseudo-transition state graph)를 만들어 모델에 입력합니다. 가상 전이상태 구조와 AlphaFold로 예측한 단백질 구조를 입력으로 받아, 아래 그림의 우측처럼 화학반응들과 단백질들 간의 점수를 예측합니다. 주어진 화학반응에 대해 점수가 가장 높은 단백질이 해당 반응을 촉매할 효소라고 예측하는 것이죠.
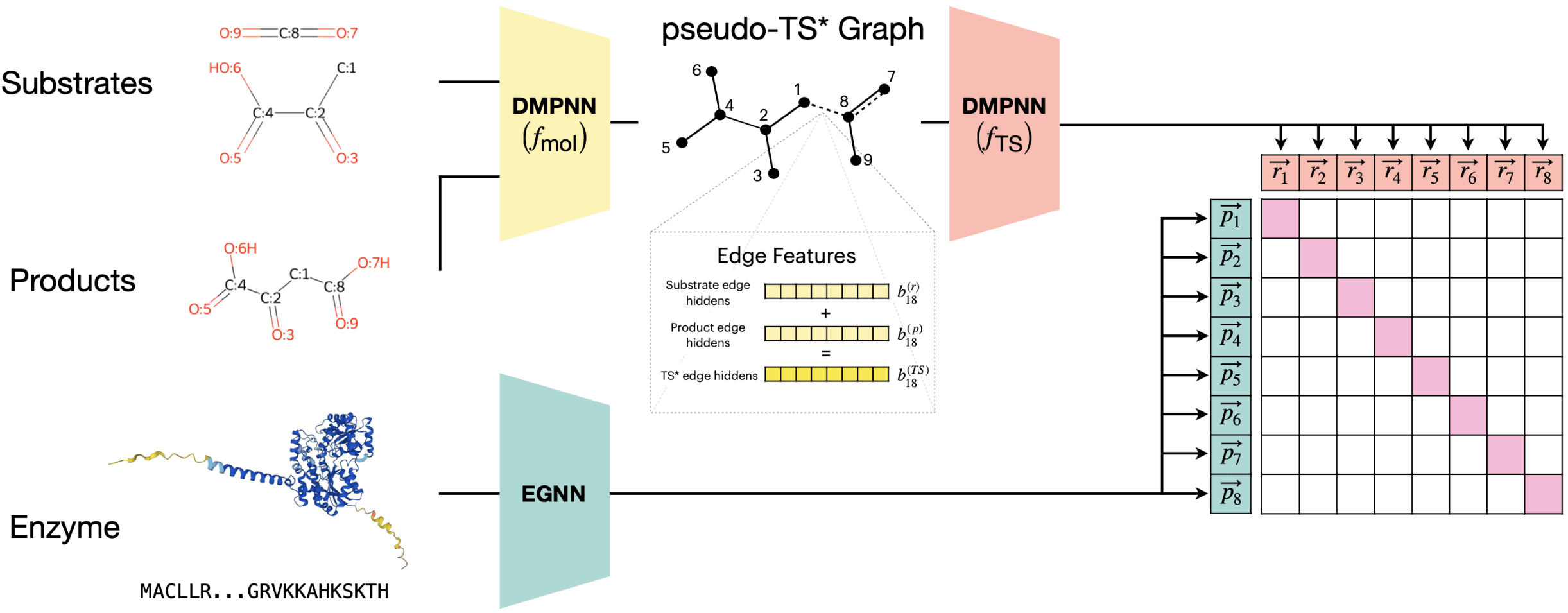
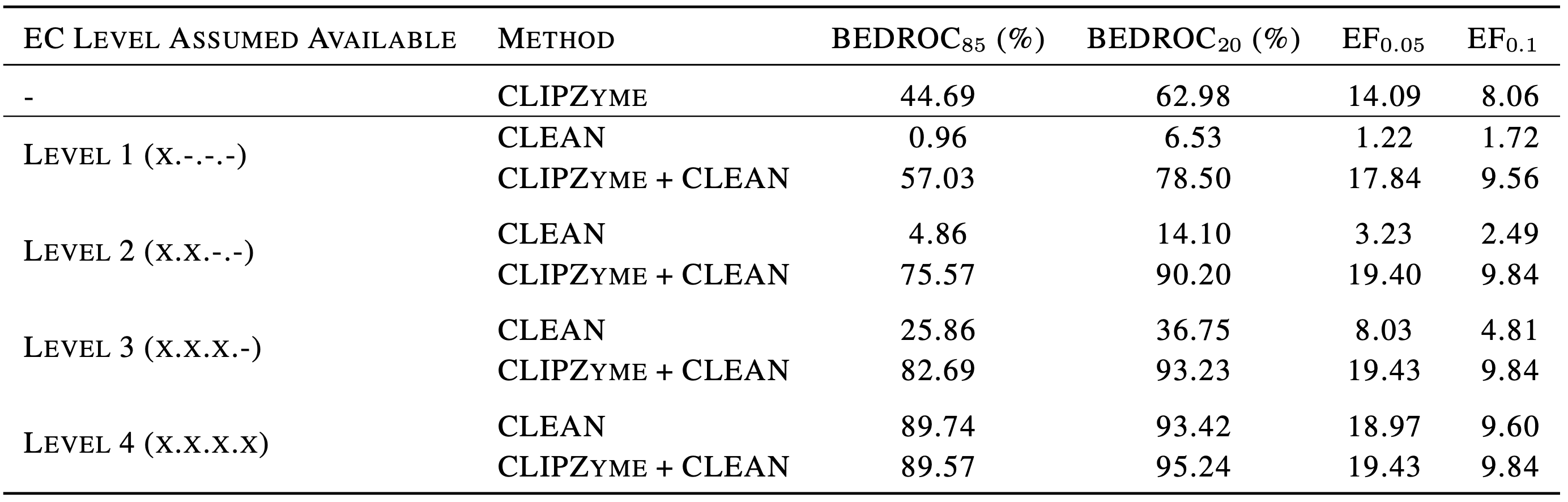
CLIPZyme의 정확도를 조사하기 위해 주어진 단백질의 EC number를 맞추는 문제에 응용한 후 CLEAN 모델과 비교했습니다. CLIPZyme 모델만으로는 주어진 단백질의 EC number를 맞출 수는 없기 때문에, 주어진 EC number로 분류되는 단백질들을 CLEAN으로 먼저 예측하고, 그 안에서 CLIPZyme이 다시 순서를 매기는 식으로 응용했습니다. 그 결과 CLEAN만을 썼을 때에 비해 CLEAN에 CLIPZyme의 예측을 함께 썼을 때 정답 효소를 찾아내는 성능이 크게 앞서는 것을 확인할 수 있었습니다.
CLEAN 같은 모델은 조사할 화학반응에 적절한 EC number를 먼저 부여해야 하지만 CLIPZyme의 경우 그런 과정 없이 임의의 화학반응을 조사할 수 있다는 장점도 가집니다.
ICML 2024 논문 미리 보기 3 - Building block의 변화와 중요도를 고려하여 분자 디자인하기
표적 단백질에 적합한 화합물을 설계하는 생성 AI 연구는 꾸준히 주목받고 있습니다. 단백질 구조가 있는 3차원 공간상에서 바로 리간드를 생성하고자 하는 시도가 많이 되고 있지만, 마치 의약화학자들이 디자인하듯 2차원 분자 구조를 생성하는 모델들도 계속해서 연구되고 있습니다.
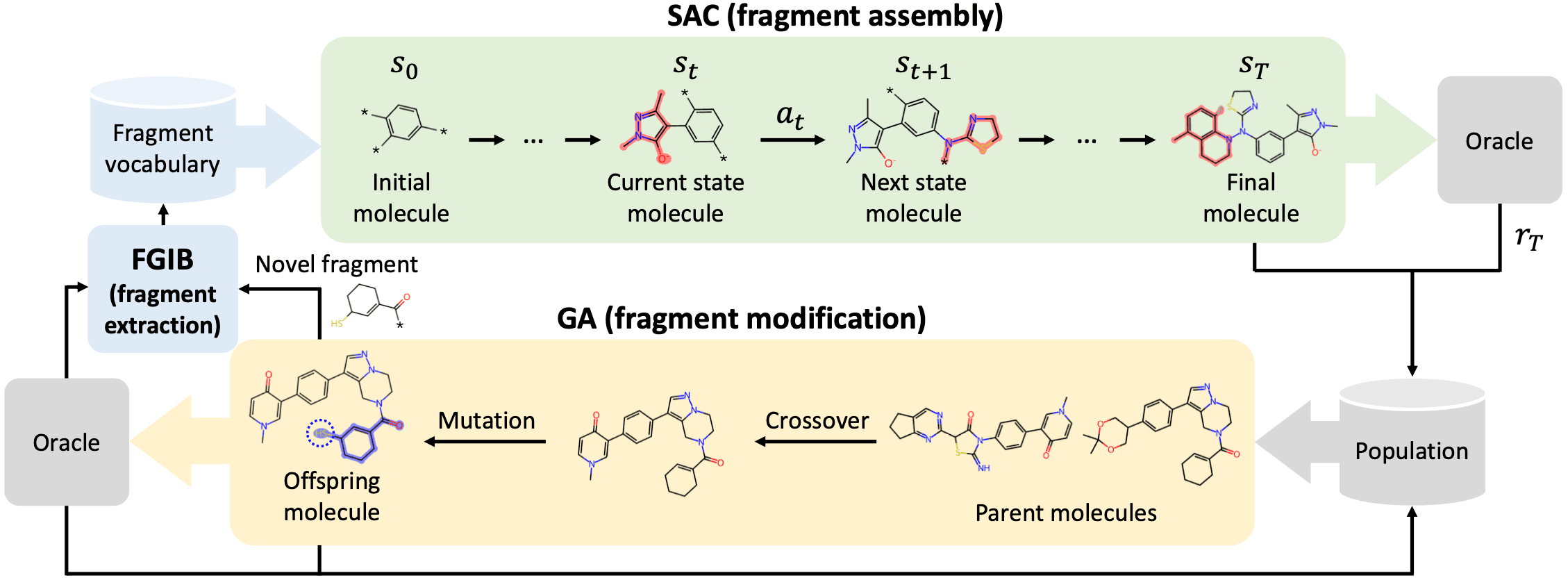
“Drug Discovery with Dynamic Goal-aware Fragments”에서 소개하는 GEAM (goal-aware fragment extraction, assembly and modification) 모델은 분자를 building block 단위로 붙여서 생성합니다. [Lee et al. ICML 2024] Building block, 혹은 fragment 단위로 분자를 생성하는 모델은 이미 여럿 있어왔는데요. GEAM은 몇 가지 매우 합리적인 차별점을 가집니다.
먼저, GEAM은 고정된 하나의 building block 집합을 사용하는 대신, 서로 다른 최적화 상황에서 목표 물성에 적합한 building block 집합을 다르게 설정하여 생성을 시작합니다. 대상 물성에 가장 크게 기여할 수 있는 building block들을 예측하고 골라 쓰는 것이죠. 또한 그렇게 처음 정한 building block들의 조합으로만 분자가 생성되는 것이 아니라, 생성 중인 분자의 부분 구조를 변형하고, 또 변형된 분자를 기반으로 building block 집합을 재설정 하여 생성을 이어나갑니다. 제목의 “goal-aware” 하고 “dynamic” 하다는 게 이러한 특성들을 말하죠. GEAM이 분자를 생성하는 과정은 아래 그림과 같습니다.
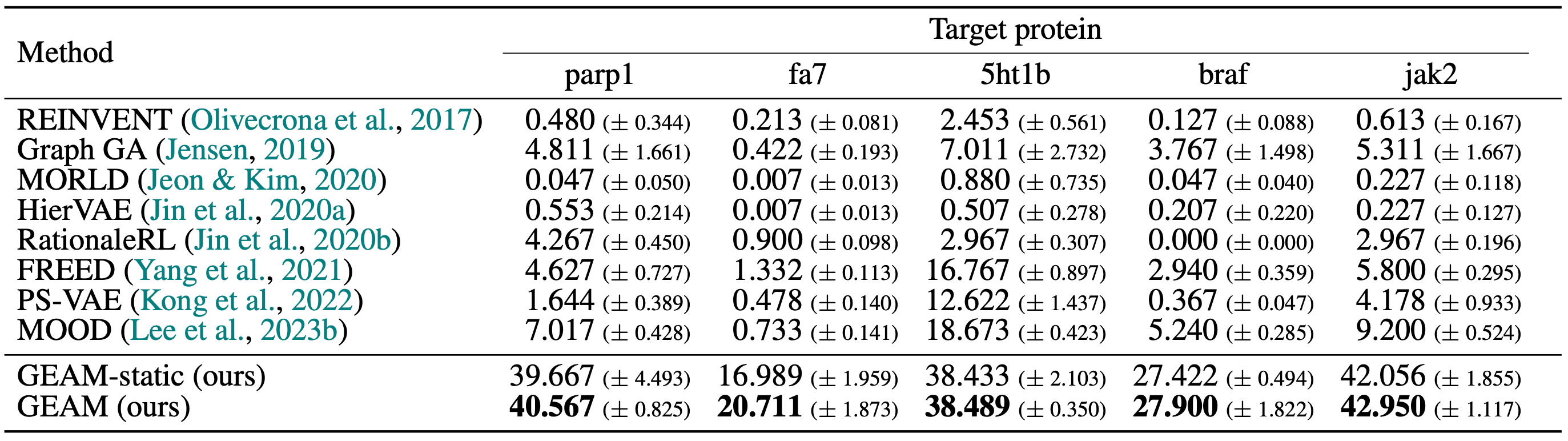
GEAM의 분자 생성 성능을 비교하기 위해 다섯 가지 표적 단백질들 (PARP1, FA7, 5-HT1B, B-Raf, JAK2)에 대해 생성한 분자들의 docking score, quantitative estimate of drug-likeness (QED), synthetic accessibility (SA) 등을 계산하여 novel hit ratio를 측정했습니다. 그 결과 비교 생성 모델들에 비해 5가지 표적에서 모두 크게 앞서는 성능을 보였습니다.
또한 GEAM이 building block을 선정하는 성능 또한 정성적으로 조사했습니다. 아래 그림은 GEAM이 생성한 분자가 표적 단백질과 상호작용 하고 있는 모습과 GEAM이 해당 분자의 각 building block이 상호작용에 기여할 것이라 예측한 정도 (weight)입니다. Weight이 크게 예측된 부위가 단백질과 특정한 상호작용을 맺고 있는 것을 확인할 수 있습니다.
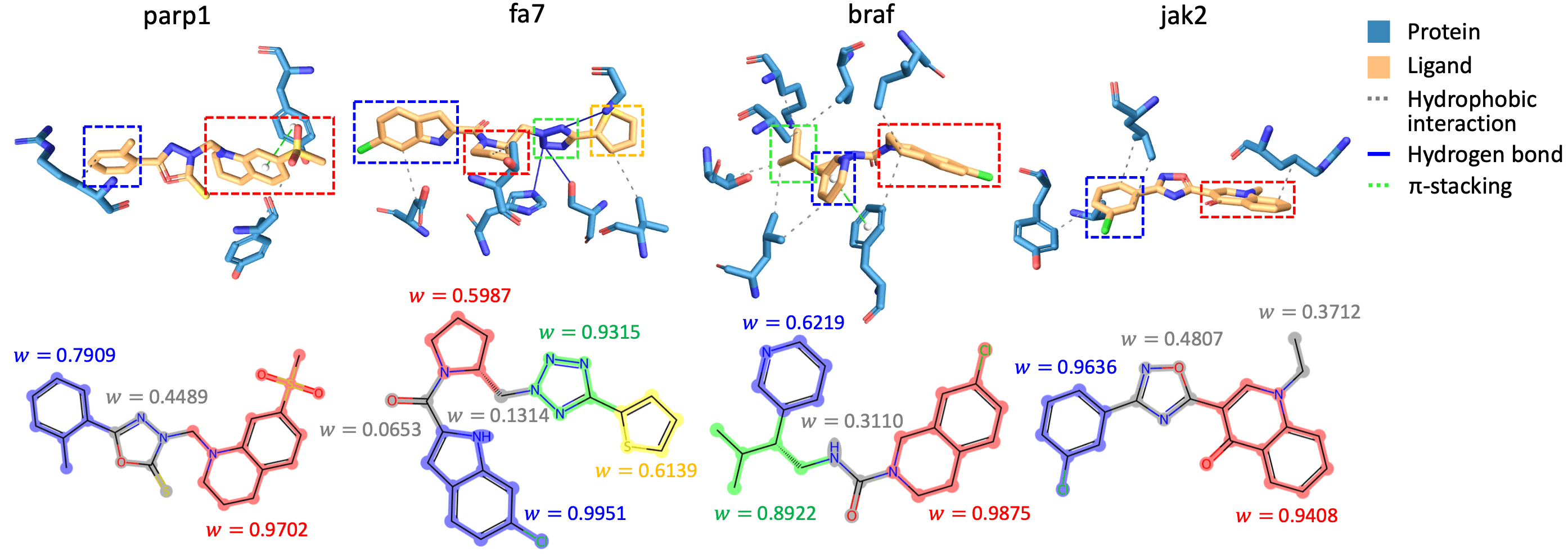
GEAM과 같은 리간드 생성 모델은 (1) 표적 단백질이 달라질 때마다 모델을 새로 학습해야 한다는 점, (2) 외부 docking 프로그램에 의존한다는 점을 제약으로 가지고 있습니다. 그럼에도 불구하고, structure–activity relationship(SAR)에 초점을 맞춰 2차원 분자 구조 디자인 성능을 강조하는, 또 다른 관점의 접근법이라고 볼 수 있겠습니다.
ICML 2024 논문 미리 보기 4 - 자연스러운 3차원 구조로 분자 디자인하기
단백질 구조가 있는 3차원 공간에서 리간드를 바로 생성하는 연구는 이번 ICML 2024에서는 많이 발표되진 않았습니다. 그만큼 문제가 어려워서일까요?
“MolCRAFT: Structure-Based Drug Design in Continuous Parameter Space”에서 소개된 MolCRAFT (Continuous paRAmeter space Facilitated molecular generaTion) 모델은 기존의 3차원 리간드 생성 모델들이 공통적으로 가지고 있던 고질적인 문제를 해결하고자 합니다. [Qu et al. ICML 2024] 그 문제란, 모델들이 자연스럽고 그럴듯한 분자를 생성하지 못한다는 점입니다.
아래 그림에서 보이는 것처럼 기존의 모델들은 부자연스런 고리 구조를 만들거나 단순한 부분구조를 반복적으로 붙이는 경향이 있었습니다. 또한 생성된 분자에 따라 올바른 결합 길이나 결합 각도를 제대로 모사하지 못하는 경우도 있었습니다.
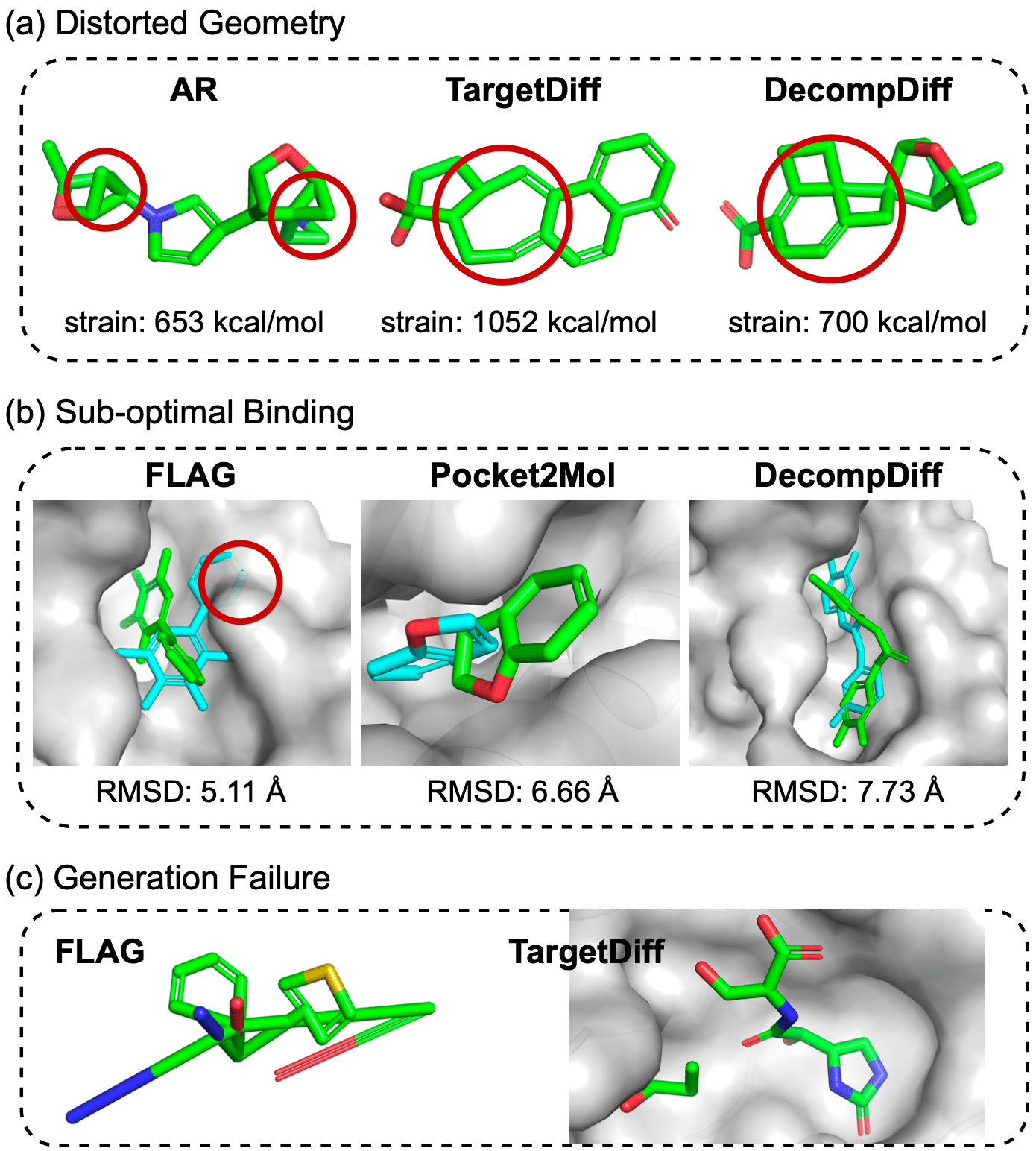
MolCRAFT는 이러한 생성 문제를 해결하기 위해 Bayesian Flow Networks(BFNs)를 사용합니다. [Graves et al. 2023] BFN은 앞선 AlphaFlow에서도 사용된 flow 기반 생성 모델의 한 종류입니다. MolCRAFT의 논문 [Qu et al. ICML 2024]에선 원자 단위 또는 fragment 단위로 순차적으로 붙여가는 자기회귀(autoregressive) 모델의 경우 단순한 부분구조만 반복 생성하는 문제를 겪기 쉽다고 분석합니다. 또한 이와 달리 확산 diffusion 기반 모델은 저해상도 이미지를 고해상도로 복원하듯 초기에 noise가 많은 원자단을 생성한 후 점차 정제 denoising 하여 분자를 만들어냅니다. 하지만 3차원 좌표처럼 연속적인 변수와 원소 종류처럼 이산적인 변수를 함께 다루는 데 어려움이 있어 이 점이 부족한 생성 성능의 원인이라고 분석합니다. 그래서 MolCRAFT는 BFN 방법을 활용해 모든 변수를 연속된 공간에서 표현할 수 있도록 설계했으며 이를 통해 기존 자기회귀 모델과 확산 모델의 단점을 동시에 극복하고자 했습니다.
논문에서는 다음처럼 다양한 결과들을 보여주는데요.
- 화학결합의 올바른 길이 분포 모사하기
- 치우침 없이 다양한 고리 구조 생성하기
- 안정적인 docking score를 가지는 분자 생성하기
- 안정적인 conformation을 가지는 분자 생성하기
여기에는 간략하게 아래의 결과만 가져와봤습니다. 임의의 표적 단백질에 대해 다양한 모델로 분자를 생성하고 그 구조를 비교한 결과입니다. 비교 모델들에 비해 MolCRAFT는 무리 에너지 strain energy가 작은 분자를 생성하고, 결합 부위에서 비교적 안정적인 포즈를 만들어내는 것을 확인할 수 있습니다.
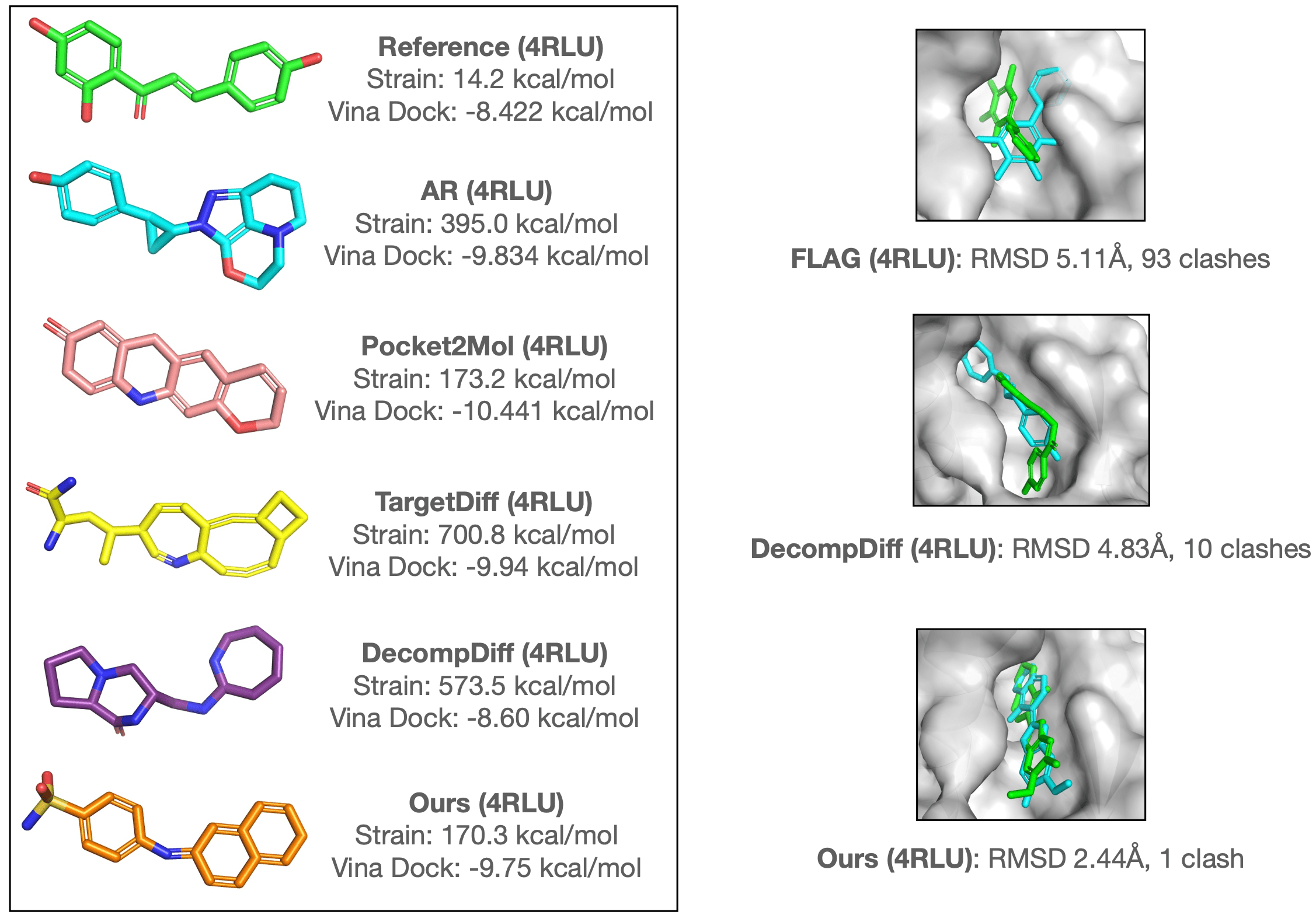
기존의 3차원 리간드 생성 모델 논문들이 통계적인 수치 성능 뒤에 숨긴 채 자세히 밝히지 않았던 위와 같은 문제를 지적하고 이를 해결하고자 한 점에서, 본 연구는 큰 의의를 가진다고 생각합니다. 다만 바로 위 그림의 결과에서 MolCRAFT의 생성물이 여전히 약간은 부자연스러운 결합각을 가지는 것을 볼 수 있는데요, 이는 저자들이 엄선한 cherry-picking 결과일 가능성이 높음에도 완벽하지 못하다는 점에서 3차원 생성 문제는 여전히 많은 발전이 필요해 보입니다.
ICML 2024 논문 미리 보기 5 - 거대언어모델로 분자 최적화하기
위의 연구 주제 통계에서 살펴봤다시피 거대언어모델을 자연과학에 적용하는 문제는 계속해서 연구자들의 관심을 받고 있습니다. 지난 ICML 2024 미리 보기에서도 이 흐름을 확인할 수 있었는데요.
이번 ICML 2024에서도 관련되는 연구들이 여럿 발표되는데요, 그중 “A Sober Look at LLMs for Material Discovery: Are They Actually Good for Bayesian Optimization Over Molecules?” 연구는 베이지안 최적화(Bayesian optimization)라는 방법으로 물질을 최적화할 때 거대언어모델을 활용하는 효과를 분석합니다. [Kristiadi et al. ICML 2024]
ChatGPT와 같은 서비스를 물질 개발에 활용하려는 시도 중 하나로, 거대언어모델에 직접 질문을 제시하는 방식이 있습니다. 이런 경우 모델이 마치 과학 원리를 이해하는 듯 보이지만 결국엔 매우 자신 있게 틀린 답을 내놓는 모습을 흔히 볼 수 있습니다.

위 같은 방식보다 간접적으로 거대언어모델을 활용하기 위해 거대언어모델을 베이지안 최적화와 함께 사용할 수 있습니다.
베이지안 최적화는 임의의 함수 ƒ(x)를 최적화 하는 일반적인 전략 중 하나인데요, 지금의 맥락에서 논항 x는 후보 분자, 함숫값 ƒ(x)는 원하는 물성으로 생각할 수 있습니다. 대부분의 분자 최적화 문제는 모든 분자 x의 물성 ƒ(x)를 일일이 알기 어려운 조건에 놓여 있죠. 베이지안 최적화는 그런 조건에서 기존에 조사된 분자들 x1, x2…과 그들의 물성 ƒ(x1), ƒ(x2)...이 주어졌을 때, 물성을 최적화하기 위해 다음에 조사해야 할 최적의 분자 후보 x*를 추정해줍니다. 이 같은 과정을 반복하면 무작위로 분자들을 조사하는 것보다 더 적은 실험 횟수로 최적의 분자를 빠르게 찾아낼 수 있게 되죠.
[Kristiadi et al. ICML 2024]에서는 베이지안 분자 최적화 문제에서 분자의 특성을 뽑아내는 데에 거대언어모델을 사용합니다. 거대언어모델은 임의의 자연어 입력을 벡터나 행렬과 같은 수치적 형태, 즉 임베딩 (embedding)으로 변환할 수 있는데요. 분자 구조를 SMILES와 같은 문자열로 표현하여 거대언어모델에 입력하면 해당 분자 구조의 임베딩을 얻어낼 수 있습니다. 마치 분자 구조를 fingerprint로 표현하는 것과 비슷하죠. 논문에서는 이러한 임베딩이 내포한 분자의 정보를 베이지안 최적화에 이용해 원하는 물성을 최적화할 때의 효율을 조사했습니다.
[Kristiadi et al. ICML 2024]에서는 다음과 같은 6가지 데이터셋과 최적화 문제에서 실험을 진행했습니다.
- Redoxmer: 흐름전지 (flow battery) 전해질의 산화-환원 전위 최소화
- Laser: 레이저의 형광 진동자 세기 (oscillator strength) 최대화
- Solvation: 흐름전지 전해질의 용매화 에너지 최소화
- Photovoltaics: 광전지 물질의 전력변환효율 (PCE) 최대화
- Kinase: Kinase 저해제의 docking score 최소화
- Photoswitches: 유기광스위치 물질의 π-π* 전이파장 최대화
먼저 사용한 거대언어모델의 종류에 따른 최적화 성능 차이를 확인했습니다. 거대언어모델로는 ChatGPT처럼 일반적인 목적으로 훈련된 모델들 T5, GPT2-M, LLaMA2-7B 등을 사용했으며, T5 모델을 화학 반응 데이터에 전이학습시킨 T5-Chem과 같은 특화 모델도 함께 비교했습니다.
그 결과 아래 그림에서 보이는 것처럼 일반적인 목적의 거대언어모델은 fingerprint처럼 단순한 정보를 사용한 경우보다도 최적화 성능이 오히려 떨어졌습니다. 반면 MolFormer, T5-Chem처럼 화학 데이터를 기반으로 (재)훈련한 모델들은 보다 빠르게 물성을 최적화하는 결과를 보였습니다.
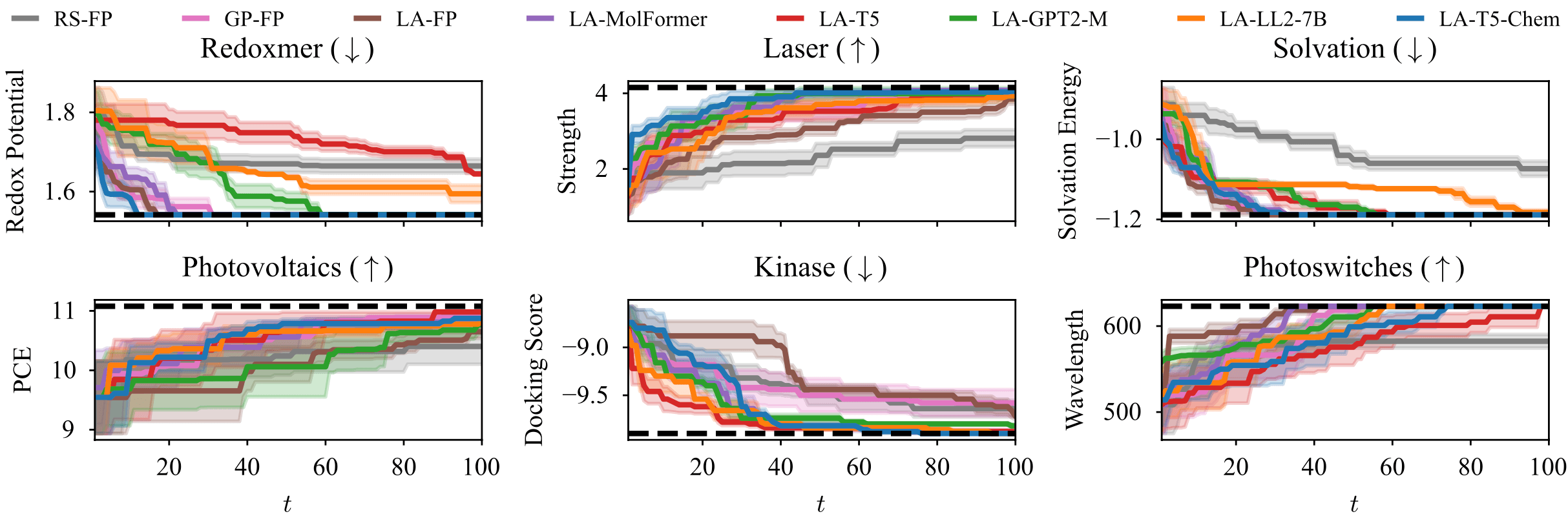
위 실험에서는 거대언어모델의 파라미터를 주어진 그대로 유지한 채 물질 정보를 임베딩했지만, 이번에는 거대언어모델에 소량의 파라미터를 새롭게 도입하고, 최적화할 물성 데이터를 기반으로 finetuning하여 비교했습니다. 아래 그림에서 실선은 거대언어모델을 그대로 사용한 경우, 점선은 finetuning을 적용한 경우를 나타내며, 대부분의 경우에 finetuning이 더 빠르게 물성을 최적화하는 모습을 볼 수 있습니다.
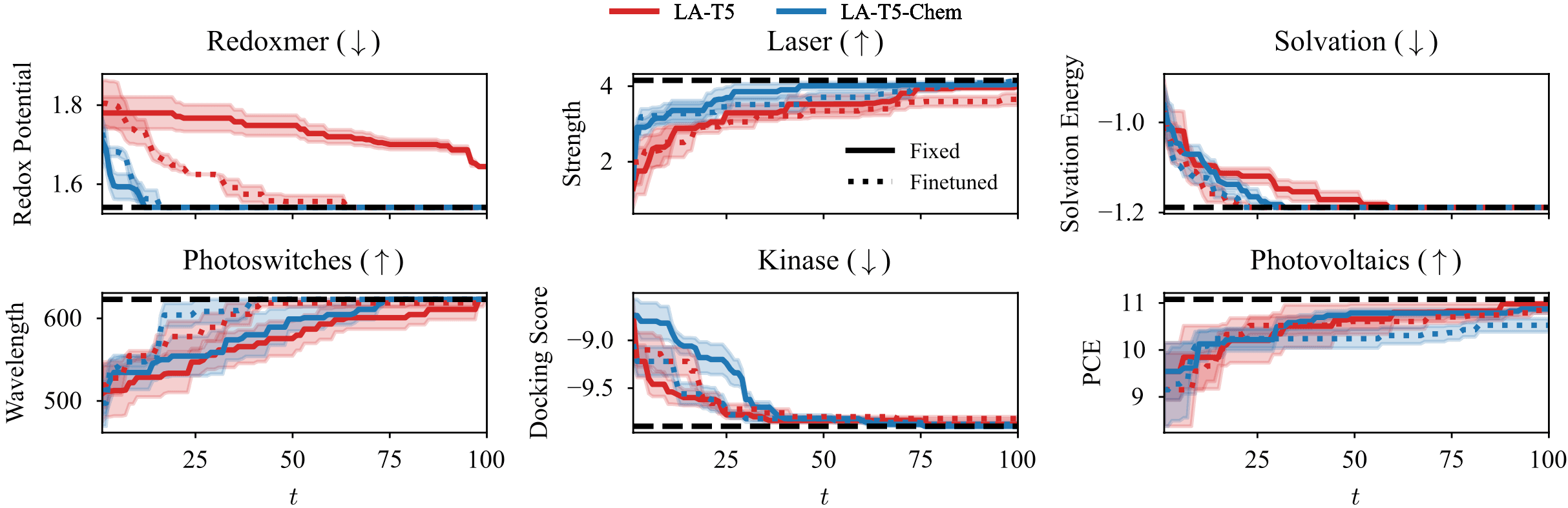
대부분의 독자들께 거대언어모델을 활용하는 방법으로는 in-context learning, 소위 프롬프트 엔지니어링 (prompt engineering)이 더 익숙할 텐데요, 실제로 in-context learning을 물질 최적화 문제에 적용한 연구도 있습니다. [Ramos et al. 2023] 이곳에 소개된 연구 [Kristiadi et al. ICML 2024]는 그와 같은 방법보다 베이지안 추정, finetuning과 같은 추가 연산이 필요하지만, 거대언어모델의 대답에 곧바로 의존하는 것에 비해 위양성 위험을 줄일 수 있을 것으로 보입니다.
ICML 2024 미리 보기를 마치며
조만간 열릴 ICML 2024에서 발표될 화학·생명과학 분야 논문들을 뽑아보고 대표적인 5가지 연구를 다뤄보았습니다.
인공지능 기반 신약개발 분야에서 최근의 가장 주목받는 소식이라면 단연 AlphaFold 3의 발표일 텐데요. [Abramson et al. Nature (2024)]ICML 2024 논문들의 경우 이미 지난 2월에 제출이 마감된 후 심사가 이뤄진 논문들이기 때문에 AlphaFold 3을 분석하거나 응용하는 연구는 아직 보이지 않습니다. 올해 말에 열릴 NeurIPS 2024도 지난 5월에 제출 마감이었던 걸 생각하면 관련 연구가 AI 컨퍼런스에 나타나는 건 적어도 내년쯤이 될 가능성이 높습니다. 하지만 arXiv나 다른 과학 저널에서는 훨씬 이른 시점에 관련 연구들이 나올 수 있겠죠.
Google이 AlphaFold 3를 통해 또 한 번 놀라운 발전을 이룬 것은 사실이지만, 앞서 살펴본 것처럼 신약개발 분야에는 여전히 풀어야 할 문제가 무궁무진합니다. 앞으로도 연구자들이 각자가 중요하게 여기는 다양한 문제를 바탕으로 흥미로운 인공지능 연구를 계속해서 이어가기를 기대합니다.