신약개발을 위한 AI 모델의 탄생


안녕하세요. HITS에서 신약개발을 위한 인공지능(AI) 모델 개발을 맡고 있는 임재창입니다.
오늘은 골격기반 분자 그래프 생성 모델(scaffold-based molecular generative model)에 대해서 설명해 드리겠습니다.
scaffold 기반 분자 생성 모델 개발 동기
분자 생성 모델은 딥러닝 기술을 이용하여 원하는 분자를 디자인하는 기술입니다. 분자 생성 모델의 태동기에는 분자 구조에 대한 사전 정보 없이 one-shot으로 한 번에 원하는 분자를 생성하려는 모델들이 주로 연구되었습니다. 이상적으로는 매력적인 접근이지만, 신약개발처럼 매우 복잡한 과제에서는 one-shot으로 분자를 디자인하는 것이 사실상 거의 불가능합니다.
대부분의 경우, 여러 번의 trial-error 과정을 거치며 점진적으로 분자 구조를 변형해가면서 원하는 물성을 만족하는 분자를 찾아갑니다. 특히 신약개발에서는 분자의 물성에 핵심적인 역할을 하는 scaffold(골격)를 고정하고, 이를 중심으로 분자의 구조를 변형시켜가며 디자인하는 경우가 많습니다.
최근에는 이러한 접근법을 반영하여, one-shot 분자 디자인보다 분자를 점진적으로 변화시켜가는 방식들이 주목받고 있습니다. 이를 위해서는 단순히 생성된 분자의 물성만을 조절하는 것이 아니라, 분자의 물성과 구조를 동시에 조절할 수 있는 분자 생성 모델이 필요합니다. 저희가 골격기반 분자 그래프 생성 모델을 논문으로 발표했을 당시에는 이러한 개념들이 아직 일반적이지 않았습니다. 하지만 저희 연구 이후, 이러한 접근이 점차 보편화되었고, 현재는 다양한 후속 모델들이 제안되며 널리 사용되고 있습니다.
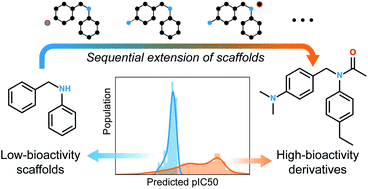
핵심 작동 원리: 생성된 분자의 scaffold를 조절하기 위한 방법
물론 생성된 분자의 분자 구조를 조절하기 위한 연구들은 이전에도 존재했습니다. 다만 기존 연구들의 주요 목적은, 생성된 분자와 기준 분자의 ‘유사도’를 조절하는 것이었습니다. 하지만 유사한 분자를 생성하는 것과, 특정 scaffold를 갖는 분자를 생성하는 것은 전혀 다른 문제입니다. 후자의 경우, 분자 구조를 보다 직접적으로 조절해야 하므로 난이도가 훨씬 더 높습니다. 기존 연구에서는 latent space 상에서 기준 분자와 생성될 분자 간의 거리를 조절함으로써 유사도를 조절하는 방식을 사용했지만, 이러한 방식으로는 생성된 분자의 scaffold를 명시적으로 고정할 수 없다는 한계가 존재했습니다.
이 문제를 해결하고자 저희는 scaffold에 원자와 화학 결합을 추가하여 분자를 디자인하는 방식을 고안하였습니다. 이 방식은 scaffold를 기반으로 분자를 확장해나가는 구조로, 생성된 분자가 항상 처음에 주어진 scaffold를 하위 구조로 포함하게 만듭니다. 이 아이디어는 SMILES 기반의 분자 생성 모델과는 잘 결합되지 않습니다. 왜냐하면 SMILES에서는 scaffold에 해당하는 부분이 반드시 연속적으로 표현되지 않기 때문입니다. 이에 저희는 분자 그래프 생성 모델을 도입하여 이 문제를 해결했습니다. 분자 그래프는 2차원 구조이기 때문에, 1차원 표현인 SMILES와 달리 기존 구조에 새로운 부분을 직접 추가하면서 분자를 디자인하는 것이 가능합니다.
모델 구조 및 algorithm
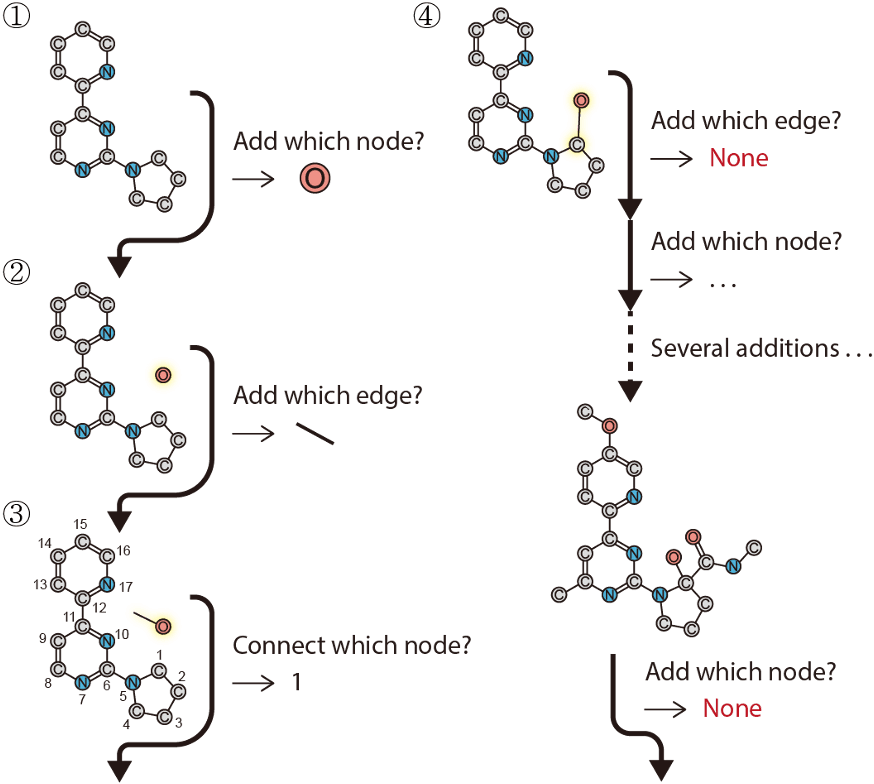
저희가 제안한 모델은 scaffold의 그래프를 입력으로 받아, 주어진 scaffold를 포함하는 분자 그래프를 output으로 생성합니다. 생성된 분자 그래프는 SMILES, SDF, MOL2 등 다양한 표현형으로 쉽게 변환할 수 있습니다. 세부 과정은 아래에 정리된 바와 같습니다. (그림2 참조)
1) 입력받은 scaffold의 그래프 생성 및 원자 feature, 화학결합 feature 초기화
2) graph neural network를 이용하여 scaffold의 원자 feature 및 화학결합 feature 업데이트
3) 새로 추가될 원자의 종류 선택 ex) C, N, O, …, T (생성종료)
4) 추가된 원자와 연결될 화학결합 종류 선택 ex) 단일결합, 이중결합, 삼중결합
5) 기존 원자들 중 추가될 원자와 연결될 원자 선택
6) 기존 분자 그래프에 원자 추가 및 원자 feature, 화학결합 feature update
7) 3~6 과정 반복. 3에서 추가될 원자 종류에 'T'가 선택되면 분자 생성 종료
모델은 '원본분자-scaffold'가 매칭되어있는 학습데이터를 학습하여 scaffold로부터 원본분자를 복원하는 방법을 학습하게 됩니다. 이러한 학습 과정을 통해 모델은 주어진 scaffold에서부터 scaffold를 하위구조로 포함하는 유효한 분자를 만드는 법을 배웁니다. 만일 생성된 분자의 물성도 조절하고 싶을 경우 학습 과정에서 scaffold뿐만 아니라 분자 성질도 input으로 받게 됩니다. '(scaffold, 물성) → 분자' 이 관계를 학습하게 되는 것이죠.
모델은 '원본 분자–scaffold'가 매칭되어 있는 학습 데이터를 기반으로, scaffold로부터 원본 분자를 복원하는 방법을 학습하게 됩니다. 이러한 학습 과정을 통해 모델은 주어진 scaffold로부터, 해당 scaffold를 하위 구조로 포함하는 유효한 분자를 생성하는 법을 익히게 됩니다. 만일 생성된 분자의 물성도 함께 조절하고 싶을 경우, 학습 과정에서 scaffold뿐만 아니라 분자의 성질 정보도 input으로 넣어줍니다. 즉, '(scaffold, 물성) → 분자'라는 관계를 학습하게 되는 것입니다.
결과
저희는 이 모델을 EGFR 저해제 개발 프로젝트에 적용했습니다. 먼저 ChEMBL에서 EGFR 관련 활성 데이터를 수집하였습니다. EGFR은 수천 개의 보고된 활성 데이터를 보유하고 있어 데이터 양이 많은 타겟에 속하지만, 수천 개 정도의 데이터만으로 딥러닝 모델의 최적 성능을 확보하기에는 여전히 부족함이 있습니다.
이를 보완하기 위해 저희는 준지도학습(semi-supervised learning)을 도입하였습니다. 준지도 학습은 label이 없는 대규모 데이터(즉, 분자 구조 정보만 있는 데이터)를 활용하여 label 부족 문제를 보완할 수 있는 방식입니다. 다시 말해, label이 없는 대규모 데이터를 통해 분자를 디자인하기 위한 화학적 규칙을 학습하고, label이 있는 데이터를 통해 EGFR에 대한 활성값을 조절하는 방향으로 모델이 학습되도록 설계한 것입니다.
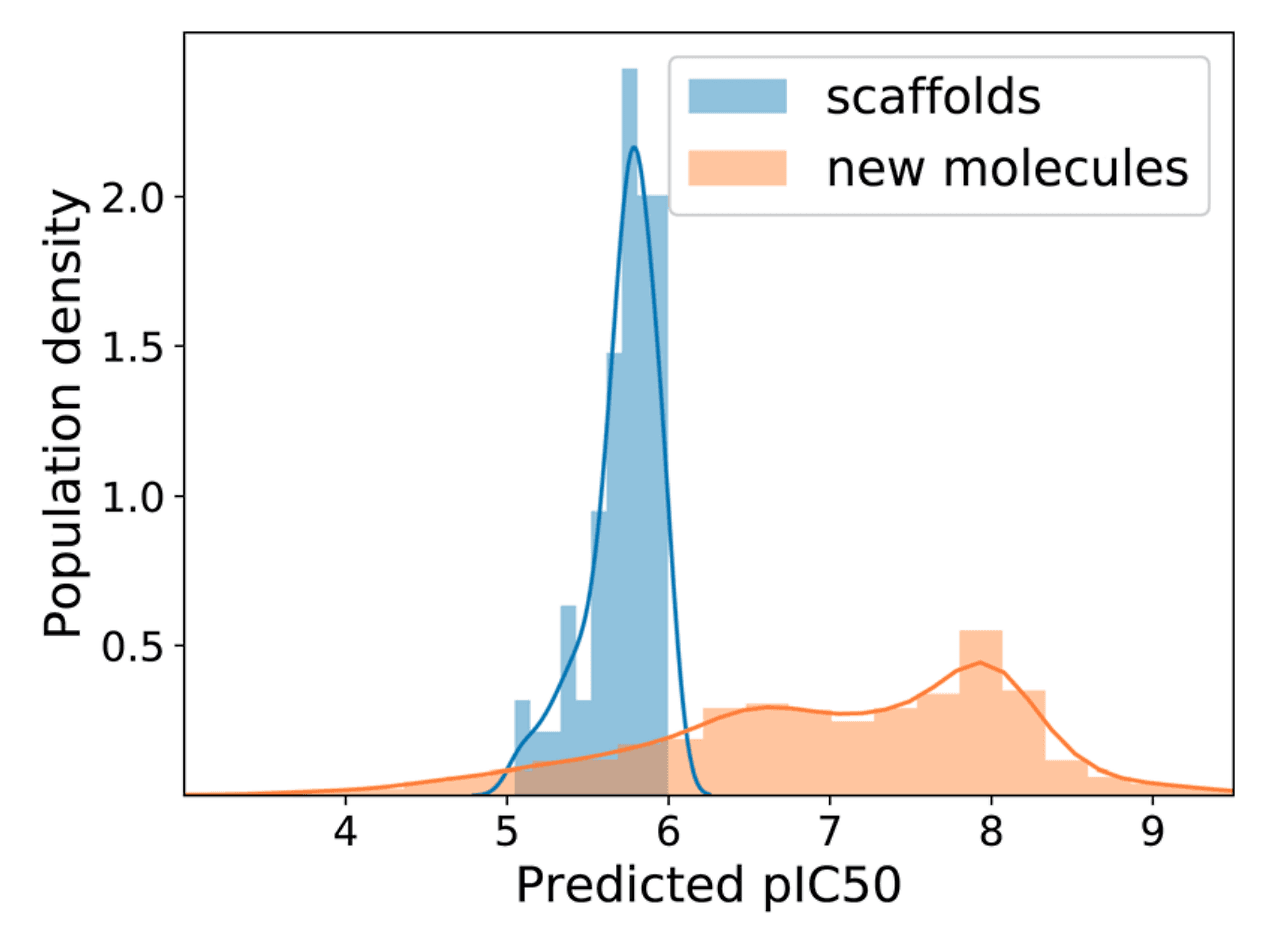
모델 학습 후에는 실제로 분자를 생성하고, 생성된 분자의 활성을 예측하여 모델의 성능을 평가했습니다. 학습에 사용되지 않은 데이터 중에서 활성값이 1μM 이하(pIC50 > 6)인 분자들의 scaffold를 추출하고, 이를 input으로 하여 새로운 분자들을 디자인하였습니다. 그 결과 생성된 분자들 중 상당수가 IC50 기준 수십 nM 수준의 활성을 가질 것으로 예측되었습니다.
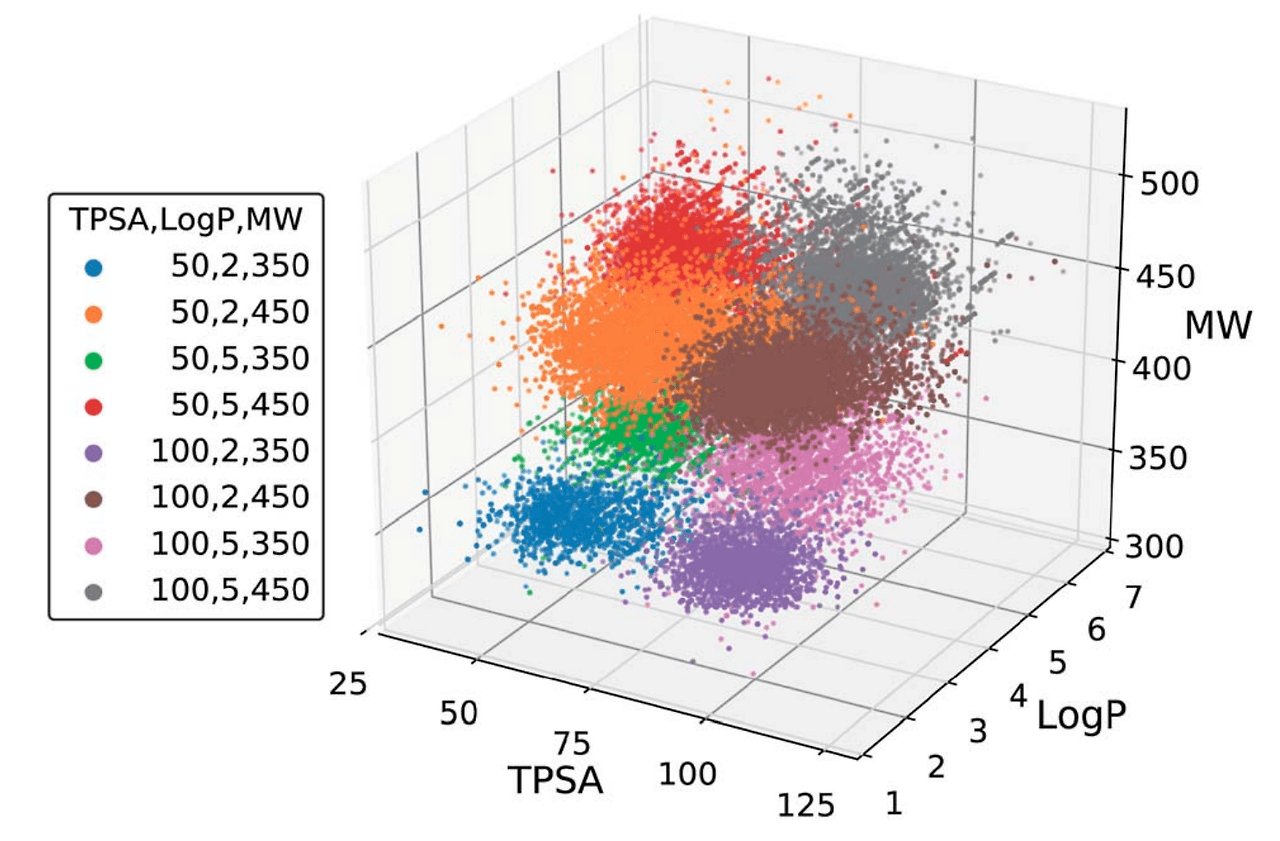
추가적으로 생성된 분자의 여러 물성을 동시에 조절할 수 있는지도 테스트해 보았습니다. 이를 위해 MW, TPSA, LogP를 조절할 수 있는 모델을 새로 학습시켰습니다. 놀랍게도 scaffold가 고정되어 있어 분자 디자인에 큰 제약이 있음에도 불구하고, 생성된 분자의 여러 물성을 동시에 조절하는 데 성공했습니다.
그림 4에서는 지정해준 물성 값에 따라 점들이 특정 영역에 모여 있는 것을 확인할 수 있는데요, 이는 생성된 분자들의 물성이 목표로 설정한 값들과 유사하게 분포하고 있다는 의미입니다. 신약 개발에서는 활성, ADME/T, solubility, permeability 등 여러 물성을 동시에 만족하는 분자를 찾는 것이 매우 중요합니다. 저희 모델이 이러한 복수의 물성을 동시에 조절할 수 있다는 것은, 실제 신약 개발에 있어 큰 잠재력을 지니고 있음을 보여주는 결과라 할 수 있습니다.
지금까지 HITS의 핵심 역량 중 하나인 분자 구조 설계와 관련하여 골격기반 분자 그래프 생성모델(scaffold-based molecular generative model)에 대해 설명해드렸습니다. 이 모델을 기반으로 저희는 LG화학과의 공동연구를 성공적으로 수행할 수 있었습니다. 이처럼 HITS는 실제 신약개발 과정에 적용되어 실질적 도움을 줄 수 있는 딥러닝 모델을 개발하고 있습니다. 이런 연구에 관심 있으시다면 HITS에 합류하세요!