Understanding Hyper Lab workflows with the FDA-approved drug Pirtobrutinib in 2023


Introduction
April 2024 was the period when HITS unveiled “Hyper Lab” to the world.
Hyper Lab was showcased at the Drug Discovery Conference (DDC) 2024, and at the American Association of Cancer Research (AACR) 2024 conference. To effectively demonstrate the use cases of Hyper Lab, we created a research scenario as an example.
In this posting, I will briefly introduce this demonstration.
Protein Setting
For the demonstration, we will be searching for a new hit candidate having similar characteristics to the BTK enzyme inhibitor pirtobrutinib, one of the FDA-approved drugs in 2023. Pirtobrutinib is a non-covalent type II inhibitor and is known to show efficacy to both wildtype and mutated BTK receptors. There are two 3D structures available in PDB, that is 8FLL and 8FLN, which are wildtype and C481S mutation structures. Here, we will use both of these structures.

Reference molecule check using Hyper Binding
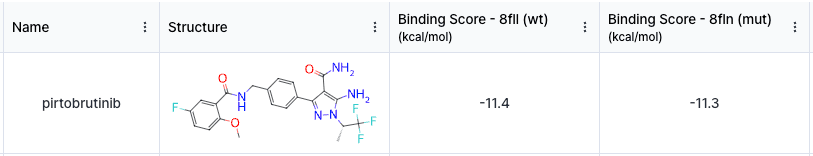
Our first goal is to check whether AI prediction values actually reflect the efficacy tendency of pirtobrutinib towards wildtype and mutation structures. To do so, we will use Hyper Binding, which is a tool for predicting drug-target binding energy. Once we calculated the values, the wildtype scored -11.4 kcal/mol while the mutation scored -11.3 kcal/mol. They both have strong binding affinities and showing similar binding tendency, just like in the literature.
 We can also check the binding pose of the ligand from the 3D Viewer. Here, we can see that the docking structure is aligned pretty well with the X-ray co-crystallized structure. We can also check the key interacting residues and the interaction types from the simplified 2D diagram.
We can also check the binding pose of the ligand from the 3D Viewer. Here, we can see that the docking structure is aligned pretty well with the X-ray co-crystallized structure. We can also check the key interacting residues and the interaction types from the simplified 2D diagram.

Virtual Screening using Hyper Screening
Since we found that the AI predictions well-reflect the experimental results, now it’s time for us to use a virtual screening tool “Hyper Screening” to find a new scaffold for the BTK receptor. For the chemical library, Hyper Lab provides some built-in libraries suitable for various scenarios, and for this case we will be using the “Kinase” library set that has 500 compounds all have a hinge binding motif. Usually this process takes about half an hour.

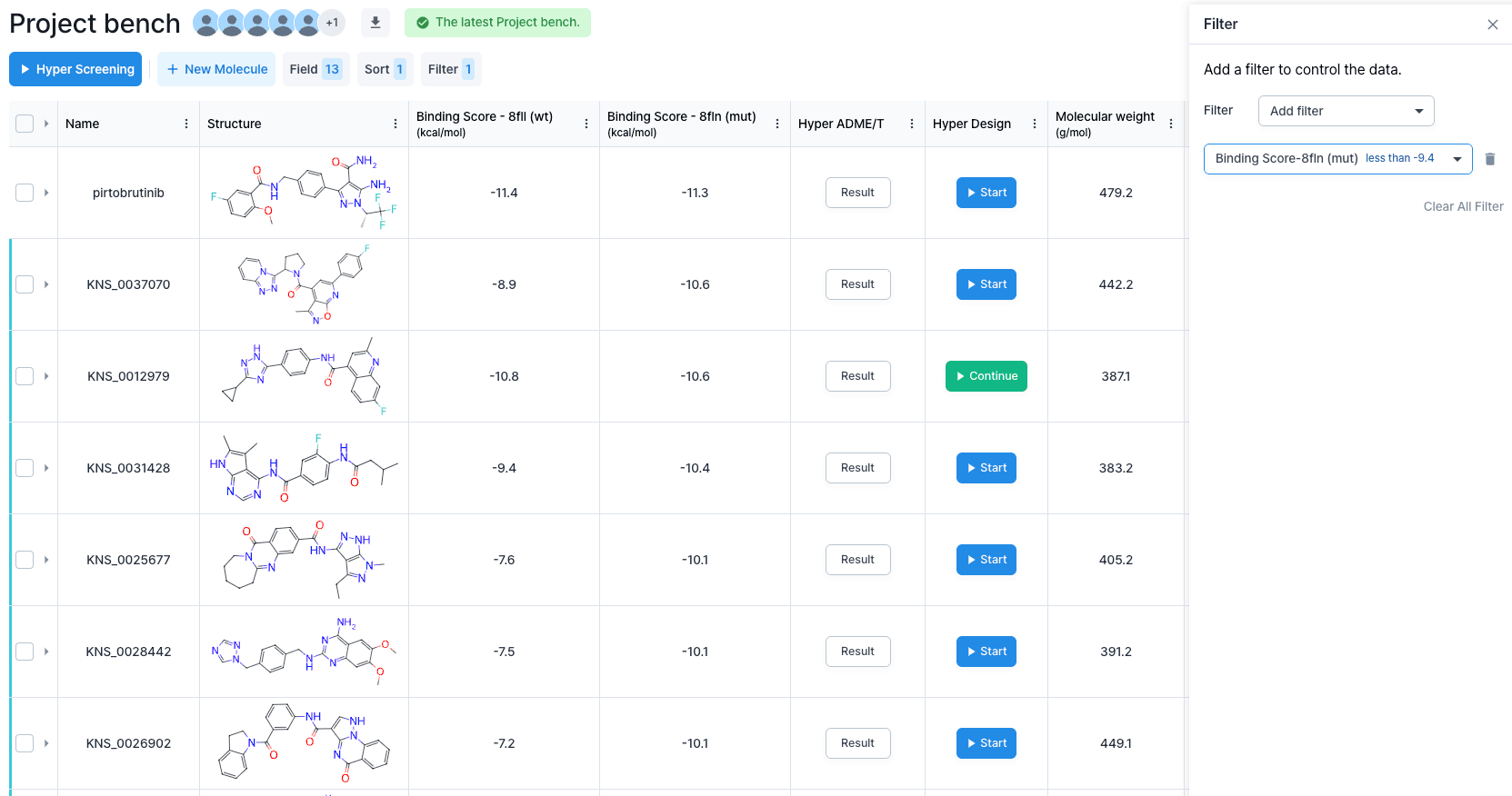
From the results, we’re going to filter out some molecules by setting a threshold based on the binding energy value. Since we’re at a screening stage, we’re not going to set our threshold value too high. Since pirtobrutinib has a binding energy value of -11.3 and -11.4 kcal/mol, we will set the threshold value to be -9.4 kcal/mol, which is 2 kcal/mol larger than those of pirtobrutinib. This allows clear off most of the compounds except for these 37 molecules.
Similar compound search
Now, we’re going to apply 2 criteria for choosing the scaffold: 1) binding energy tendency similar to that of pirtobrutinib, and 2) compound novelty. For the first criterion, we see that the best binding score molecule has -10.6 kcal/mol in the mut., but the value drops to -8.9 kcal/mol in the wt. structure. This does not follow the tendency of pirtobrutinib, so we move on to the second highest scored molecule. It seems like the second best scored molecule scored -10.8 kcal/mol in wt and -10.6 kcal/mol in mut. Now, for the second criterion, novelty, we will search for the similar compounds based on the chemical structure similarity. It seems like the 90% similarity molecules don’t have any patents, so we will choose this structure as the scaffold and begin to develop by using the molecular design tool “Hyper Design.”

ADME/T Prediction using Hyper ADME/T
To see if there is any room for improvement, let’s check the ADME/T properties. The “Hyper ADME/T” tool allows you to predict various types of ADME/T & physicochemical values. And here, our scaffold value shows the log P value to be 4.6, and moderate solubility, but an unstable metabolic stability. Therefore, in Hyper Design, we will design the molecule to improve the metabolic stability as well as improved binding affinity.

Molecule design using Hyper Design
So, using our Hyper Design tool, we can directly edit the structure of a molecule, but we can also let AI generate and recommend us a list of candidates by either adding or replacing a functional groups of a molecule. Here, we will use the ”replace” function to stabilize the metabolic stability by reducing the log P value.
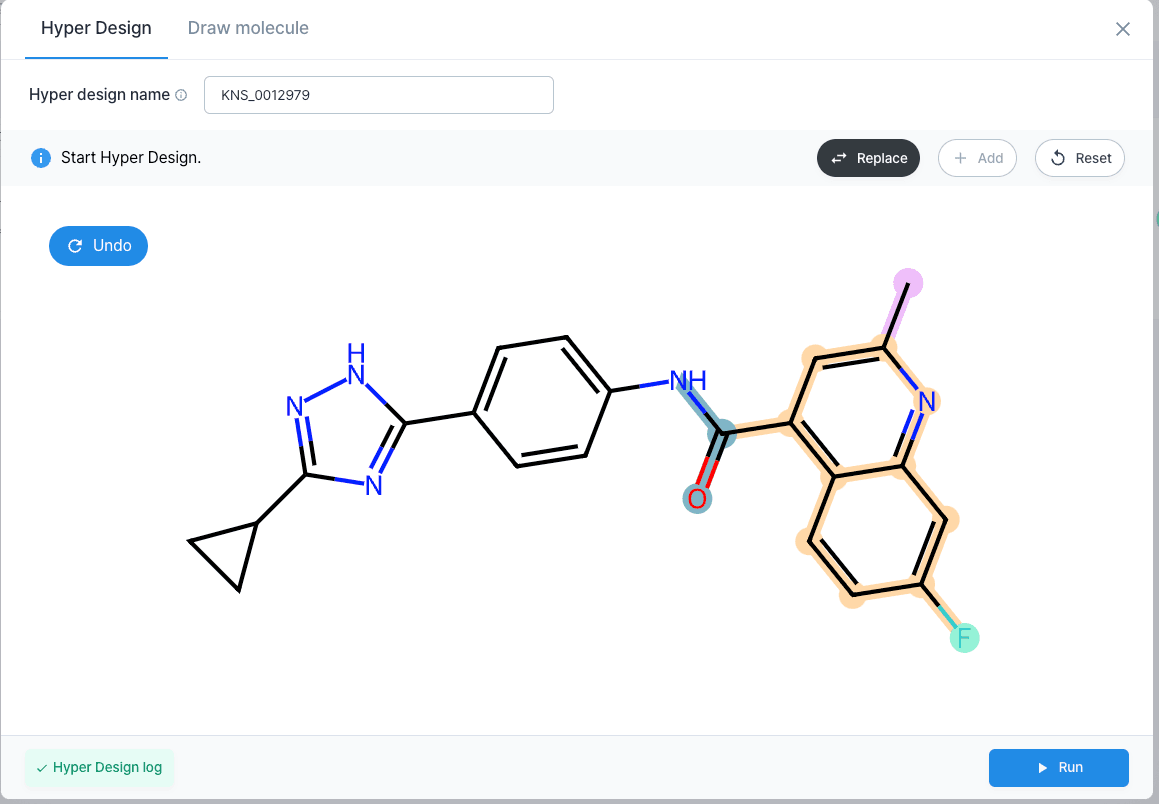
First, we will let AI recommend us derivates by replacing the core. From the recommended list, we chose the 3rd candidate which has a lower log P value as well as having a higher binding energy. As a result, the log P value decreased, leading to an increase in polarity and stabilizing the metabolic stability.
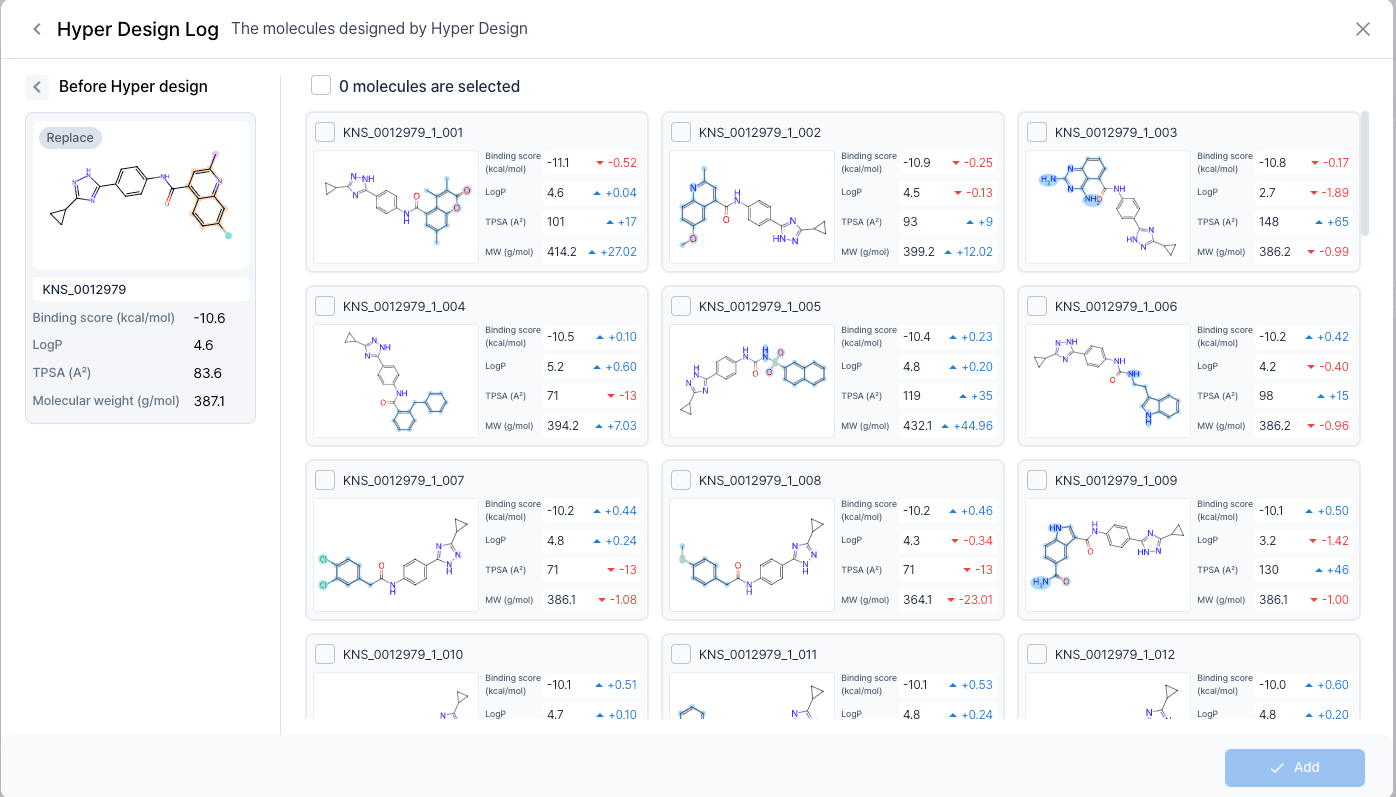
We also checked if this molecule binds to the mut. structure, and found that it also binds to the mut. in a stable manner. By checking the similar chemical compounds, we also found out that it also has some novelty. Therefore, we chose this molecule as our final candidate for the actual assay.

Wrapping up
As you can see in the demonstration above, Hyper Lab is a platform that helps you easily conduct your drug development research. With the latest AI technology incorporated into the platform, Hyper Lab offers accurate predictions as well as fast computation speeds. We encourage you to try out the Hyper Lab platform. If you have any further questions about the demo, please refer to the upcoming demo video tutorials that will be uploaded soon.