How AlphaFold 3 and Advanced Docking Are Revolutionizing Drug Discovery


Why Co-folding Matters After AlphaFold 3
In 2021, Nature published AlphaFold 2, transforming structural biology by predicting protein 3D structures from amino acid sequences with near-atomic accuracy, outperforming existing methods in CASP14.
In May 2024, AlphaFold 3 took a further leap, enabling direct prediction of complex “co-folded” structures—including proteins, nucleic acids (DNA/RNA), small-molecule ligands, ions, and post-translational modifications (PTMs)—using a probabilistic diffusion-based architecture. This means it can simultaneously model how a target protein subtly adapts when a ligand binds, all within a single model.
This shift moves drug discovery from the traditional “fitting ligands into rigid proteins” docking paradigm to a “co-folding” paradigm, where interacting molecules are folded together. This has significant implications for both initial design and structure-based optimization.
This article examines the limitations of traditional docking methods, showcases how HyperLab’s Hyper Binding Co-folding enhances real-world drug discovery workflows, and presents case studies illustrating its transformative impact. For a quick overview, watch the video below.
Limitations of Traditional Docking Methods
1. Difficulty Capturing Protein Dynamics
Most docking software assumes proteins, composed of thousands of atoms, are rigid structures. In reality, proteins shift by a few angstroms before and after ligand binding, and these subtle changes significantly affect binding poses and scores. While molecular dynamics (MD) simulations can model flexibility, they are complex to set up and exponentially slower, requiring thousands of times more computation.
2. Dependence on Predefined Binding Sites

To reduce computational load, docking typically requires specifying binding sites in advance. However, without a specified site, accuracy often drops sharply (e.g., from ~50% to below 20%), posing a frequent challenge.
3. Challenges in Selecting PDB Structures
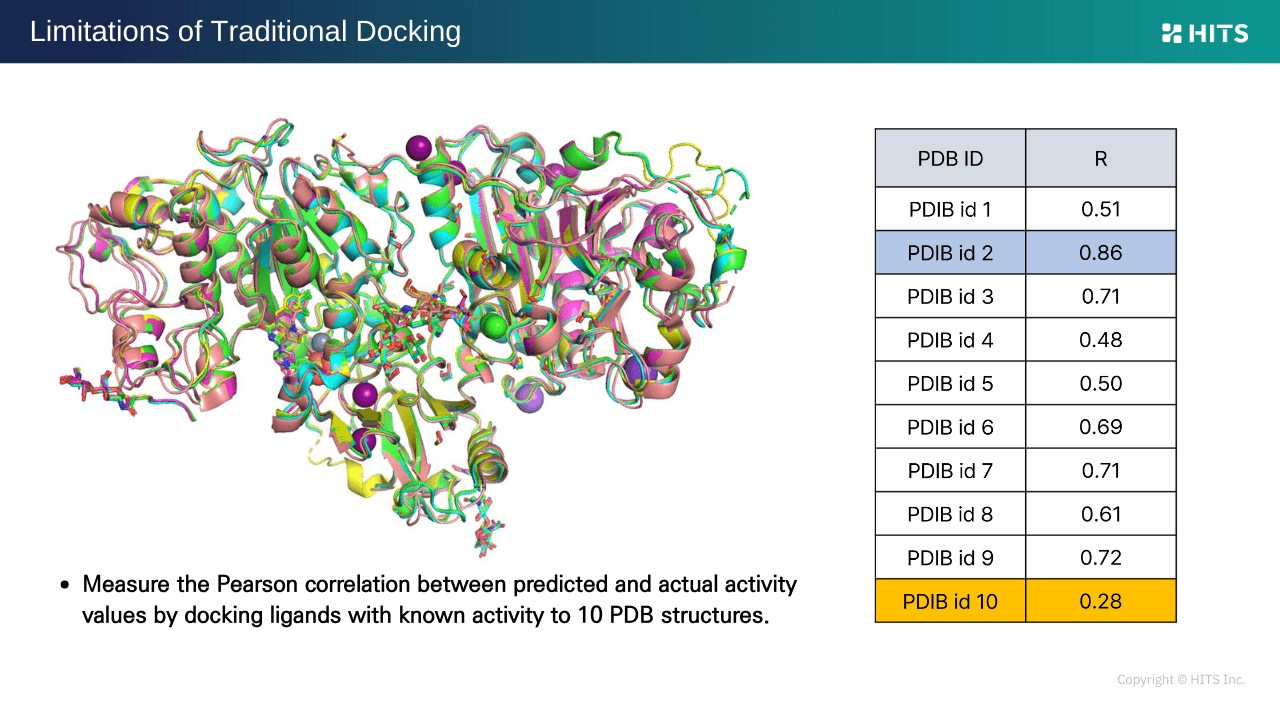
When multiple PDB structures exist for a target, the selection of a structure significantly affects prediction accuracy. For the same derivative set, Pearson correlation coefficients can range from 0.86 to as low as 0.28, depending on the model. Issues like missing 3D structures, low-quality homology models, or suboptimal reported holo structures further reduce accuracy, especially for first-in-class projects targeting novel mechanisms.
Overcoming Traditional Docking Limitations with Hyper Binding Co-folding
1. No PDB Required: Sequence-Based Binding Prediction
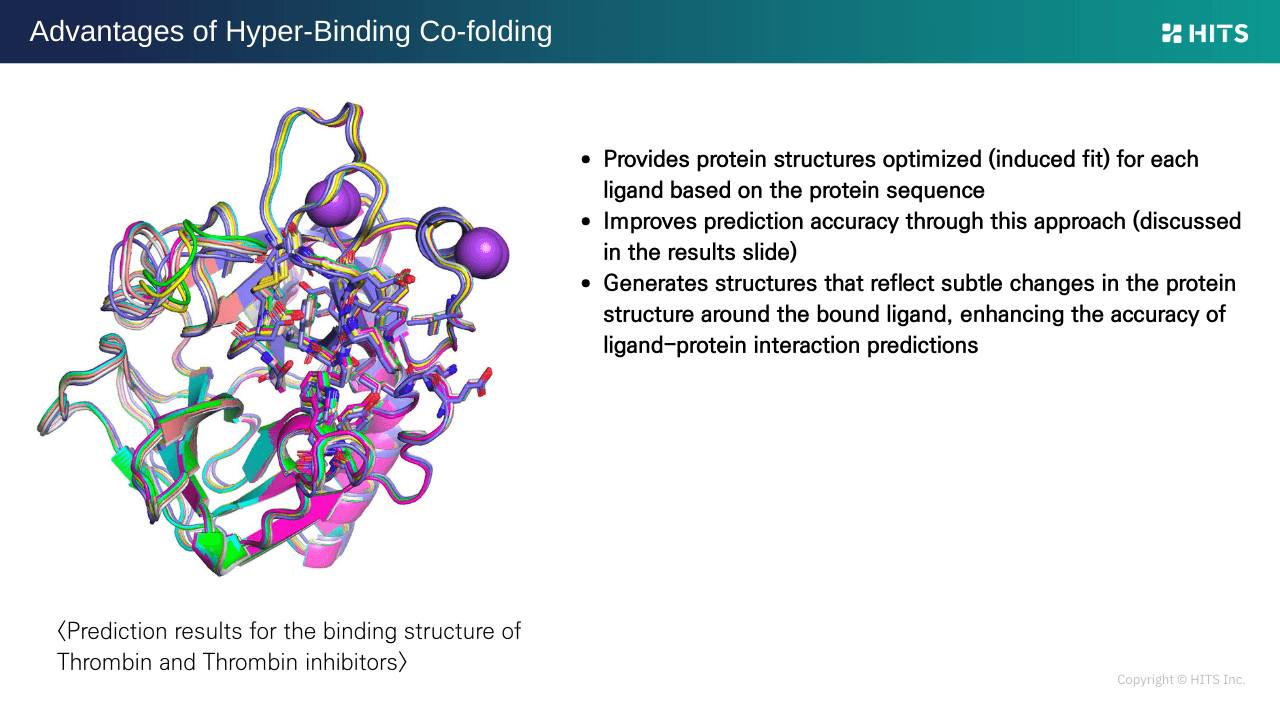
Unlike traditional docking, which forces ligands into rigid protein structures, Hyper Binding Co-folding predicts ligand-induced protein conformational changes. It can generate binding poses from protein sequences alone, even without PDB structures, making it valuable from the earliest stages of drug discovery.
2. Flexibility in finding ite Selection
Hyper Binding Co-folding produces valid poses without requiring predefined binding sites. When prior knowledge (e.g., hinge binding) is available, incorporating it can further enhance prediction precision.
3. Automated Structure Selection
Traditional docking requires selecting the most suitable PDB structure, but Co-folding eliminates this step by generating structures directly from sequences. In some cases, sequence-based structures from Co-folding have outperformed carefully selected holo structures in predicting activity.
4. User-Friendly Workflow
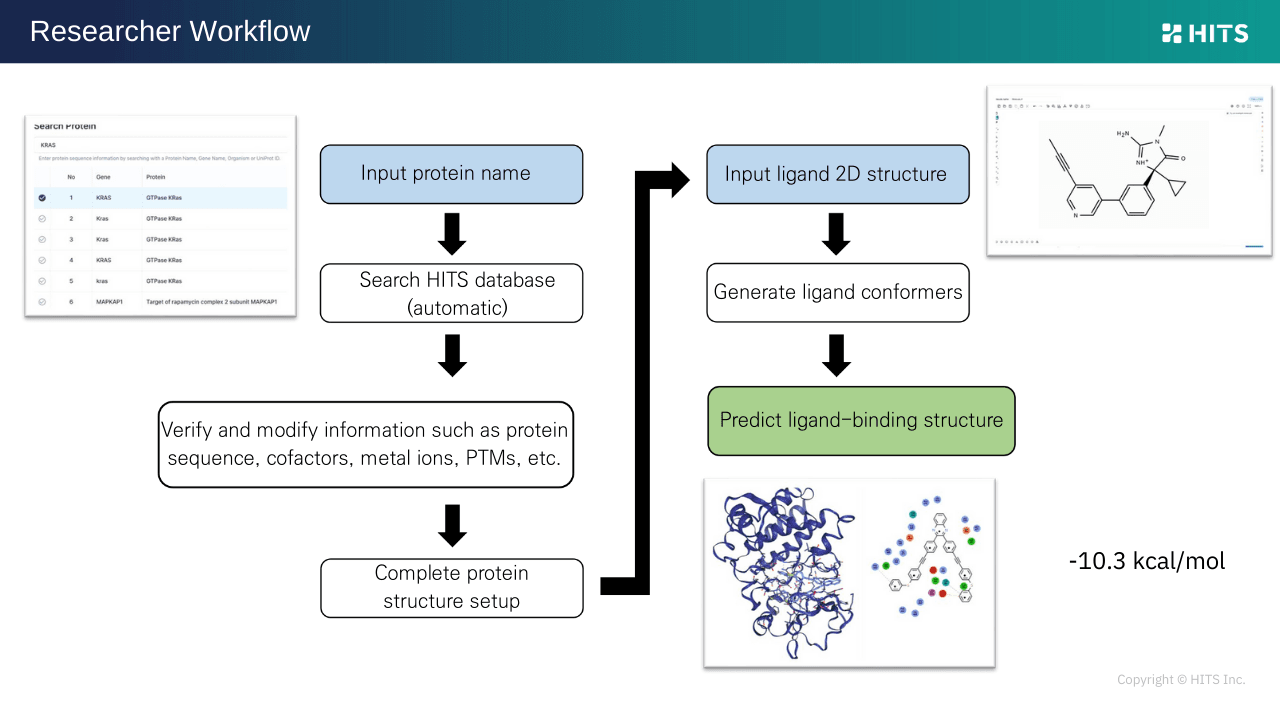
HyperLab simplifies workflows: input a protein name (e.g., EGFR, KRAS, BTK), and it automatically suggests UniProt data, cofactors, ions, and PTMs. Ligands can be drawn or uploaded, with results displayed instantly as 3D poses, 2D interaction maps, and predicted energies. Designed for experimental researchers, HyperLab requires no steep learning curve, significantly boosting workflow efficiency.
Results and Case Studies
Case 1: JAK2 Derivative Set
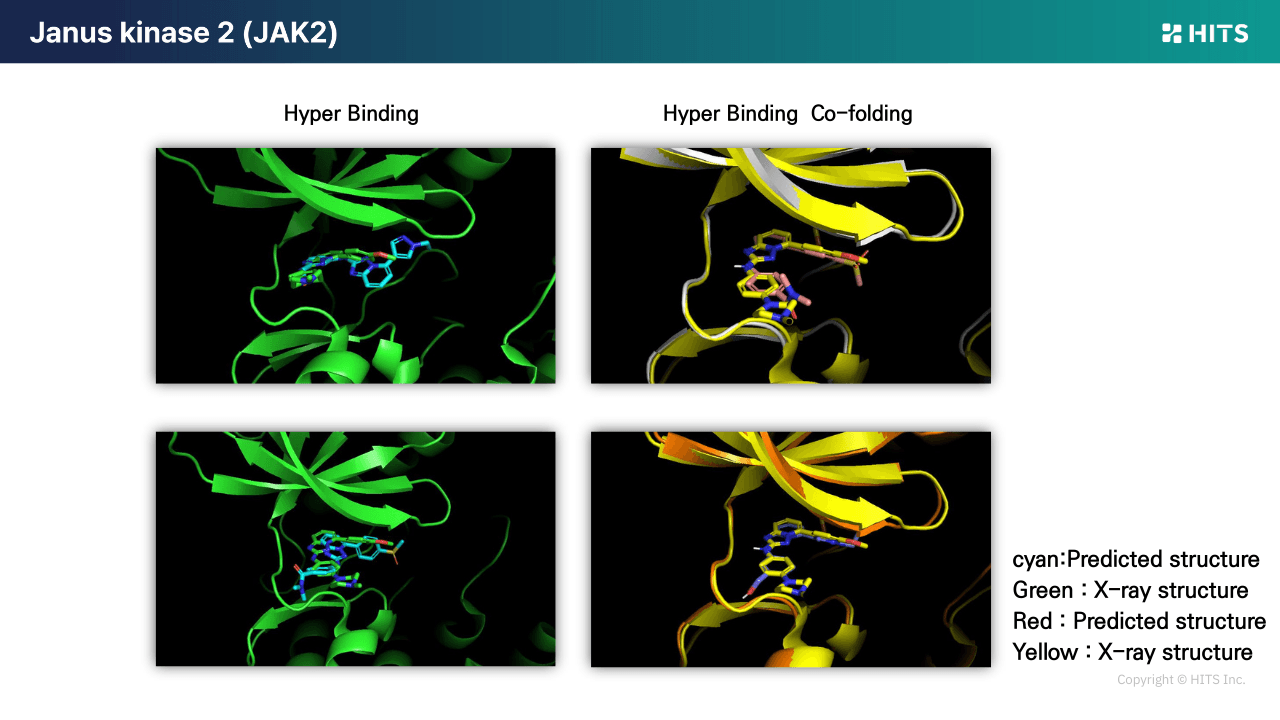
Traditional docking achieved a correlation coefficient of ~0.38 for activity prediction, limiting its utility for prioritizing synthesis. In contrast, Hyper Binding Co-folding raised the correlation to 0.71, enabling practical differentiation between candidates worth synthesizing and those to skip. It also significantly improved X-ray pose accuracy.
Case 2: Molecular Glue Complexes
For E3 ligase–target–glue ternary complexes, Co-folding predictions closely matched experimental structures. Notably, removing the glue altered the predicted binding mode, demonstrating that the model captures glue-induced surface changes not just numerically but structurally.
Benchmarking Hyper Binding Co-folding Performance
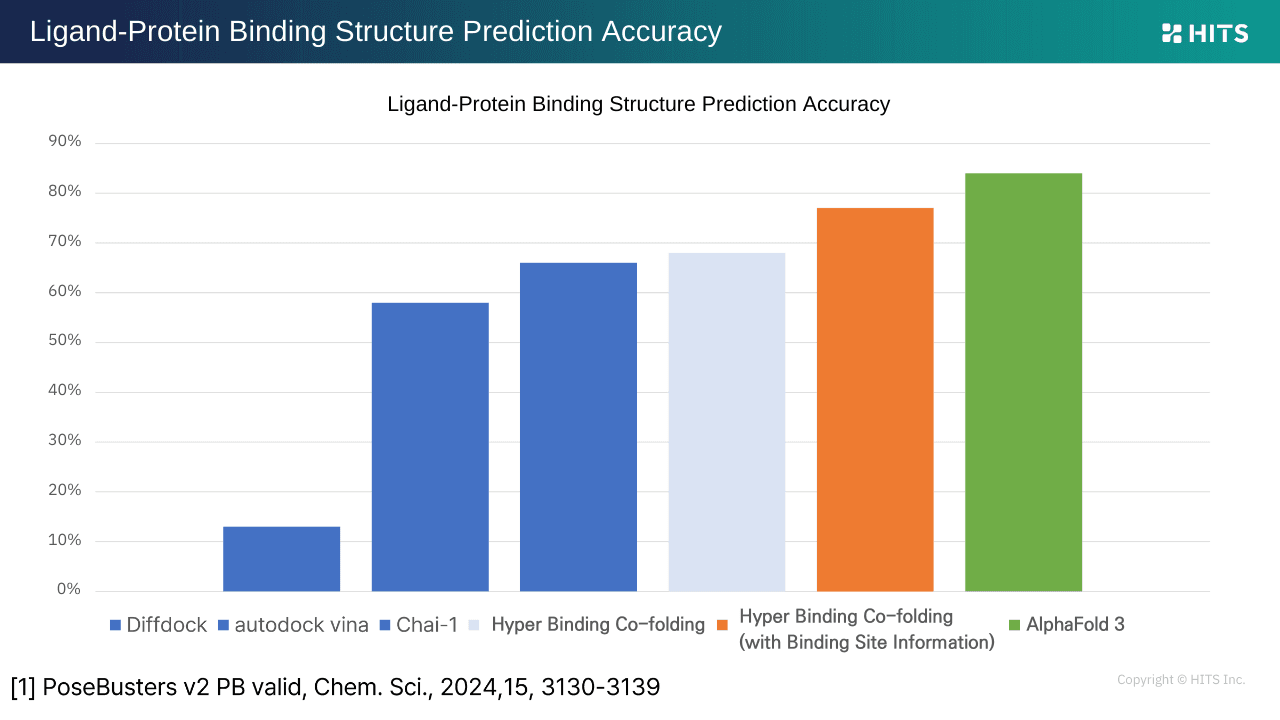
According to PoseBusters, AlphaFold 3 achieved the highest binding pose prediction accuracy at ~86%, with Hyper Binding Co-folding following at ~76%, surpassing tools such as DiffDock, AutoDock Vina, and Kai (Co-folding). Remarkably, Hyper Binding Co-folding achieved this using only protein sequences and binding sites. In activity prediction, traditional docking scored below 0.1, enhanced traditional docking (Hyper Binding) reached ~0.35, and Co-folding performed even better, delivering results closer to experimental decision-making needs.
Limitations of Co-folding
Co-folding is not a silver bullet. In rare cases with few similar PDB structures, predictions may falter, and data-driven models occasionally mispredict chirality (though post-processing can mitigate this). Capturing large-scale conformational transitions remains challenging, but recent efforts to integrate MD trajectory data into training are showing promise in addressing this.
More accurate and faster — even without PDB
In summary, Hyper Binding Co-folding predicts ligand and protein interactions together, accounting for induced fit, generates poses from sequences without PDBs, and reduces the burden of binding site specification. Benchmarks and projects demonstrate improved pose accuracy and activity prediction, enabling faster, more efficient decisions in early drug discovery.
🚀Try Hyper Binding Co-folding for Free!
Register your target now and try it yourself!
AI-Powered Drug Discovery Platform: HyperLab
- [Free Trial] https://buly.kr/Csk8wuI
- [Contact Us for Implementation] https://abit.ly/6tr1uz